
CH Activation Hot Paper
Consecutive b,b
’
-Selective C(sp3)H Silylation of Tertiary Amines with
Dihydrosilanes Catalyzed by B(C6F5)3
Huaquan Fang, Kaixue Xie, Sebastian Kemper, and Martin Oestreich*
Dedicated to Professor Siegfried Blechert on the occasion of his 75th birthday
Abstract: Tris(pentafluorophenyl)borane has been found to
catalyze the two-fold C(sp3)H silylation of various trialkyl-
amine derivatives with dihydrosilanes, furnishing the corre-
sponding 4-silapiperidines in decent yields. The multi-step
reaction cascade involves amine-to-enamine dehydrogenation
at two alkyl residues and two electrophilic silylation reactions
of those enamines, one inter- and one intramolecular.
Selective functionalization of C(sp3)H bonds is an impor-
tant goal in synthetic chemistry.[1] One way to achieve this is
by transition-metal-catalyzed C(sp3)H silylation,[2,3] and
recently selected boron Lewis acids also emerged as catalysts
for this purpose.[4] For example, B(C6F5)3has been shown to
abstract hydride from a-C(sp3)H bonds of amines to result in
the formation of iminium ions and the borohydride;[5] that
iminium ion is CH acidic and can be deprotonated by
another molecule of the amine, affording the corresponding
enamine along with the ammonium borohydride[6,7]
(Scheme 1, gray box). The net reaction is a dehydrogenation
that enables subsequent bond formation with electrophiles in
the b-position to the nitrogen atom, thereby representing
a formal activation of the b-C(sp3)H bond. This process has
already been employed for silylation,[8] alkylation,[9] deutera-
tion,[10] and olefination[11] of the b-carbon atom of various
(a)cyclic tertiary amines (Scheme 1, top). Of note, Park and
Chang merged the C(sp3)H silylation with a B(C6F5)3-
catalyzed intramolecular Friedel–Crafts-type silylation[12] for
the synthesis of bridged silicon-containing nitrogen hetero-
cycles starting from N-arylated piperidines.[8a] However, the
undirected silylation of acyclic tertiary amines[3c] as well as
their challenging two-fold C(sp3)H silylation are unprece-
dented. We disclose here a b,b’-selective C(sp3)H silylation
of acyclic tertiary amines and dihydrosilanes catalyzed by
B(C6F5)3to directly arrive at sila analogues of piperidines
(Scheme 1, bottom left). These are valuable building blocks in
medicinal chemistry,[13] for example, for the dopamine
receptor antagonist sila-haloperidol (Scheme 1, bottom
right).[14] Different from our approach, established syntheses
typically start from divinyl-substituted silanes employing
a sequence of hydrobromination or hydroboration–oxida-
tion–sulfonylation followed by dialkylation of a primary
amine.[15]
We began our investigation with optimizing the two-fold
C(sp3)H silylation of benzyldiethylamine (1a!3aa;
Table 1). Treatment of 1a and Ph2SiH2(2a, 2.0 equiv) with
20 mol% of B(C6F5)3in p-xylene at 150
8
C afforded 3aaafter
15 h in 56% yield (Table 1, entry 1). Previous reports had
indicated that the use of a metal oxide[8a] or a silyl triflate[5c] as
an additive could improve the reactivity.[16] However, sub-
stoichiometric amounts of CaO or SrO decreased the yield
(Table 1, entries 2 and 3). The addition of 40 mol% of a silyl
triflate improved the reactivity (Table 1, entries 4–6), and
a 75% yield of 3aa was obtained with Me3SiOTf as the
additive. That yield was somewhat lower when using less and
more Me3SiOTf, respectively (Table 1, entries 7 and 8). The
reaction was completed within 2 h while a further shortened
Scheme 1. B(C6F5)3-catalyzed b-C(sp3)H functionalization of tertiary
amines. R groups=various aryl and alkyl groups as well as H;
Ar=aryl group.
[*] Dr. H. Fang, K. Xie, Dr. S. Kemper, Prof. Dr. M. Oestreich
Institut fr Chemie, Technische Universitt Berlin
Strasse des 17. Juni 115, 10623 Berlin (Germany)
E-mail: [email protected]
Homepage: http://www.organometallics.tu-berlin.de
Supporting information and the ORCID identification number(s) for
the author(s) of this article can be found under:
https://doi.org/10.1002/anie.202016664.
2021 The Authors. Angewandte Chemie International Edition
published by Wiley-VCH GmbH. This is an open access article under
the terms of the Creative Commons Attribution License, which
permits use, distribution and reproduction in any medium, provided
the original work is properly cited.
A
ngewandte
Chemi
e
Communications
How to cite: Angew. Chem. Int. Ed. 2021,60, 8542–8546
International Edition: doi.org/10.1002/anie.202016664
German Edition: doi.org/10.1002/ange.202016664
8542 2021 The Authors. Angewandte Chemie International Edition published by Wiley-VCH GmbH Angew. Chem. Int. Ed. 2021,60, 8542 –8546

reaction time to 1 h resulted in a lower yield (Table 1,
entries 9 and 10). Other arene solvents were tested but none
provided a better outcome (Table 1, entries 11–13). A control
experiment verified that Me3SiOTf is unable to mediate the
reaction in the absence of B(C6F5)3(Table 1, entry 14). Less
B(C6F5)3or Ph2SiH2(2a) as well as lowering the temperature
to 120
8
C led to a decreased reactivity (Table 1, entries 15–17).
The volume of the reaction vessel was also examined, and the
results indicate that vessels smaller than 10 mL are detrimen-
tal (Table 1, entries 18 and 19). We ascribe this to catalyst
inhibition by dihydrogen at high pressure.[7] A good yield was
restored on a 5.0 mmol scale when performing the two-fold
C(sp3)H silylation in an open system with a continuous flow
of nitrogen gas (Table 1, entry 20).
We continued exploring the scope under the optimized
reaction setup (Scheme 2; cf. Table 1, entry 9). It must be
noted that reductive C(sp3)N bond cleavage[17] is competing
in any of the reactions summarized in Scheme 2, and
secondary amines are the major byproducts (not quantified
because of their volatility). N-Benzylated diethylamine
derivatives bearing various electron-donating or-withdrawing
substituents on the aryl moiety reacted with Ph2SiH2(2a)to
furnish the corresponding 4-silapiperidines in moderate to
good yields (1b–l!3ba–la; gray box). All halo groups (1g–j)
and a trifluoromethyl group (1k) were compatible. Tertiary
amine 1l containing a methyl ether underwent demethyla-
tion/silylation, and the free phenol was isolated in 50% yield
after purification by flash chromatography on silica gel (1l!
3la). A lower yield was obtained for a naphth-2-ylmethyl
instead of the benzyl group (1m!3ma). The bis(4-silapiper-
idine) 3na was formed in 47% yield by four-fold C(sp3)H
silylation of 1n. Replacing the benzyl group by an alkyl group
was feasible (1o-q!3oa-qa). Notably, the two-fold C(sp3)H
silylation of substrate 1o bearing two ethyl groups and one
cyclohexyl group proceeded chemoselectively at the ethyl
groups to form 3oa. Substituted 4-silapiperidine derivatives
were obtained from tertiary amines with groups other than
ethyl (1r–t!3ra–ta). As expected, 1t gave 3ta with essen-
tially no diastereoselectivity (cis/trans =58:42). Attempted
but failed cyclizations included tertiary benzylamines as
precursors having two isopropyl, cyclohexyl, isobutyl, or
phenethyl groups as well as 1-benzylazepane (see the
Supporting Information for details).
We also tried the silylation of the tertiary aniline
derivative 1u which did not react under Parks and Changs
catalytic system (Scheme 3).[8a] Bicyclic 4uaand tricyclic 5ua
formed in yields of 50% and 8%, respectively. The propor-
tion of 5ua increased at longer reactions times, for example,
44% yield of 4uaand 25% yield of 5uaafter 3 h. As for the
aforementioned method,[8a] intramolecular Friedel–Crafts
C(sp2)H silylation[12] is favored over intramolecular
C(sp3)H silylation.
Table 1: Selected examples of the optimization of B(C6F5)3-catalyzed two-
fold C(sp3)H silylation.[a]
Entry Additive (mol%) Solvent t[h] Yield [%][b]
1– p-xylene 15 56
2 CaO (50) p-xylene 15 48
3 SrO (50) p-xylene 15 50
4Me
3SiOTf (40) p-xylene 15 75
5tBuMe2SiOTf (40) p-xylene 15 66
6iPr3SiOTf (40) p-xylene 15 62
7Me
3SiOTf (20) p-xylene 15 67
8Me
3SiOTf (80) p-xylene 15 60
9Me
3SiOTf (40) p-xylene 2 75 (73)
10 Me3SiOTf (40) p-xylene 1 42
11 Me3SiOTf (40) toluene 2 74
12 Me3SiOTf (40) benzene 2 62
13 Me3SiOTf (40) C6H5Cl 2 55
14[c] Me3SiOTf (40) p-xylene 2 0
15[d] Me3SiOTf (40) p-xylene 2 49
16[e] Me3SiOTf (40) p-xylene 2 68
17[f] Me3SiOTf (40) p-xylene 15 61
18[g] Me3SiOTf (40) p-xylene 2 60
19[h] Me3SiOTf (40) p-xylene 2 34
20[i,j] Me3SiOTf (40) p-xylene 12 (65)
[a] All reactions were performed on a 0.050 mmol scale in a 10 mL sealed
tube. [b] Yields determined by 1H NMR spectroscopy using mesitylene
as an internal standard; isolated yields in parentheses. [c] Without
B(C6F5)3. [d] 10 mol% B(C6F5)3used. [e] 1.5 equiv Ph2SiH2(2a) used.
[f] Run at 120
8
C. [g] 5.0 mL sealed tube used. [h] 1.0 mL sealed tube
used. [i] Open system with a continuous flow of nitrogen gas.
[j] 5.0 mmol scale.
Scheme 2. Scope I: Variation of the tertiary amine. Reaction conditions
(0.10 mmol scale): B(C6F5)3(20 mol%), Me3SiOTf (40 mol%), Ph2SiH2
(2a, 2.0 equiv), and p-xylene (0.80 mL) at 150
8
C for 2 h. Yields are
isolated yields. [a] See Table 1, entry 20. [b] Starting from N-ethyl-N-(3-
methoxybenzyl)ethanamine (1l). [c] 40 mol% of B(C6F5)3, 80 mol% of
Me3SiOTf, and 4.0 equiv of Ph2SiH2(2a) used. Bn=benzyl, Cy=cyclo-
hexyl.
A
ngewandte
Chemi
e
Communications
8543Angew. Chem. Int. Ed. 2021,60, 8542 –8546 2021 The Authors. Angewandte Chemie International Edition published by Wiley-VCH GmbH www.angewandte.org

We next assessed the dihydrosilane scope in the reaction
of model substrate 1a(Table 2). Diarylsilanes 2b–eexhibited
good reactivity, furnishing the corresponding products in the
same yield range as compared to 2a (1a!3ab–ae; Table 2,
entries 1–4). No reaction was seen with sterically hindered
dimesitylsilane (2f; Table 2, entry 5). Modest yield was
obtained with MePhSiH2(2g in 1a!3ag; Table 2, entry 6)
but the synthesis of a spirocyclic derivative with 1-silaindane
(2h) was low yielding (Table 2, entry 7).[18] The dialkylsilane
Et2SiH2(2i) afforded desired 3aiin moderate yield (Table 2,
entry 8), but again, there was no reaction with bulky tBu2SiH2
(2j; Table 2, entry 9). The reaction of the primary hydrosilane
PhSiH3yielded only trace amounts of the 4-silapiperidine
(not shown).
The benzyl group in 4-silapiperidines such as 3aaserves as
a linchpin for further manipulations (Scheme 4). Debenzyla-
tion was achieved by treatment with 1-chloroethyl chlorofor-
mate followed by the reaction of the resulting carbamate with
MeOH (3aa!6). The benzyl group can also be converted
into a benzoyl group by oxidation with KMnO4in the
presence of BnNEt3Cl (3aa!7).
To gain insight into the reaction mechanism of this two-
fold C(sp3)H silylation, deuterium-labeling experiments and
stoichiometric experiments were performed (Scheme 5). The
reaction of 1awith Ph2SiD2(2a-d2) under standard conditions
gave 3aa-d3in the expected yield with 41% deuterium
incorporation in the benzylic position as well as at the
acarbon atoms (Scheme 5, top). This result confirms the
known reversible hydride abstraction from C(sp3)H bonds
ato an amine nitrogen atom.[6] Importantly, 6% deuterium
incorporation was also detected for the bcarbon atoms, which
is evidence for hydrogenation of the enamine intermediate. In
the case of diethyl-substituted 1a, silylation is faster than this
backward reaction. Conversely, di-n-butyl-substituted 1v
shows a different outcome (Scheme 5, top). None of the
hypothetical 4-silapiperidine 3va-d3was found (not shown)
but instead 1v-d3with the usual deuteration in the a-
positions. However, the deuteration grade in the b-positions
was 24%, demonstrating that enamine hydrogenation is now
a competitive if not the only reaction pathway for more
hindered alkyl chains. To inspect the influence of the
Me3SiOTf additive, we mixed 1a and B(C6F5)3in an
equimolar ratio (Scheme 5, bottom). This known reaction[6]
led to the formation of the three boron species 8a–10a in
52%, 30%, and 17% yield, respectively, and this product
distribution was not affected by the addition of 1.0 equiv of
Me3SiOTf.
On the basis of the above experimental results and the
literature precedent[6,8] as well as DFT calculations by Park
and Dang,[8b] a plausible reaction mechanism is proposed
(Scheme 6). B(C6F5)3promotes hydride abstraction from the
tertiary amine 1a to generate the iminium borohydrides 11a
Scheme 3. Consecutive C(sp3)H/C(sp2)H silylation of an aniline
derivative.
Table 2: Scope II: Variation of the hydrosilane.[a]
Entry Hydrosilane R R’Yield [%][b]
12b 4-MeC6H44-MeC6H465 (3ab)
22c 4-tBuC6H44-tBuC6H465 (3ac)
32d 4-FC6H44-FC6H467 (3ad)
42e Ph Naphth-1-yl 68 (3ae)
52f Mes Mes no reaction (3af)
62g Ph Me 40 (3ag)
72h 1-silaindan-1,1-diyl traces (3ah)
82i Et Et 42 (3ai)
92j tBu tBu no reaction (3aj)
[a] Reaction conditions (0.10 mmol scale): B(C6F5)3(20 mol%),
Me3SiOTf (40 mol%), hydrosilane 2(2.0 equiv), and p-xylene (0.80 mL)
at 150
8
C for 2 h. [b] Isolated yield. Mes=mesityl.
Scheme 4. Elaboration of an N-benzylated 4-silapiperidine. [a] 1) 1-
chloroethyl chloroformate (1.2 equiv), CH2Cl2,0
8
CtoD, 1 h; RT, 20 h;
2) MeOH, D, 1 h; [b] KMnO4(3.0 equiv), BnNEt3Cl (3.0 equiv), CH2Cl2,
D,3h.
Scheme 5. Deuterium-labeling and stoichiometric experiments. Individ-
ual deuteration grades were estimated by 1H NMR spectroscopy. The
overall deuteration grades of 2.87 D for 3aa-d3and 2.98 D for 1v-d3
were determined by mass spectrometry.
A
ngewandte
Chemi
e
Communications
8544 www.angewandte.org 2021 The Authors. Angewandte Chemie International Edition published by Wiley-VCH GmbH Angew. Chem. Int. Ed. 2021,60, 8542 –8546
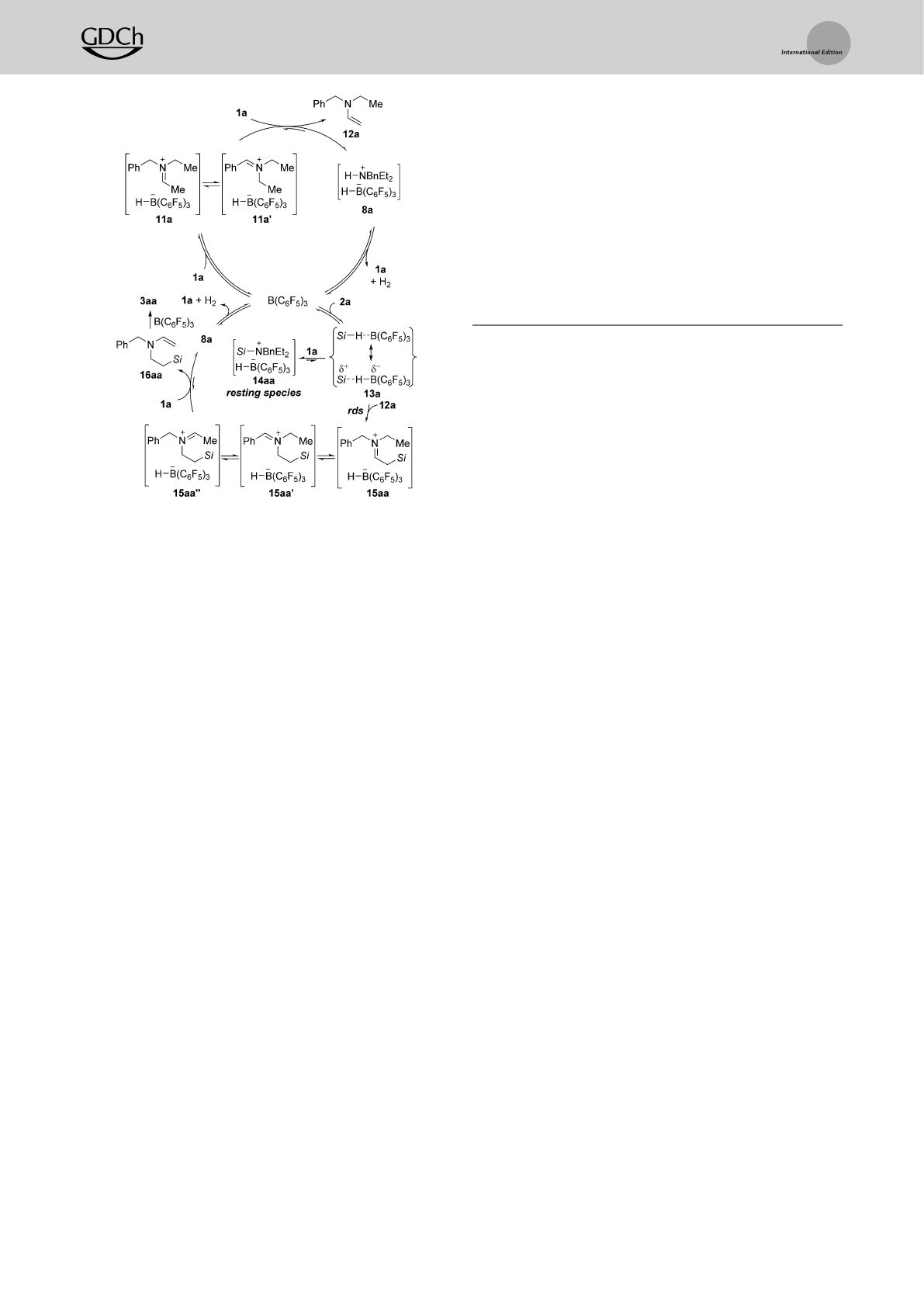
and 11a
’
in equilibrium. Their subsequent deprotonation by
unreacted 1a yields enamine 12a and FLP-type dihydrogen
adduct 8a; these can regenerate the free amine 1a and the
catalyst B(C6F5)3along with release of dihydrogen.[7,19] The
thus-formed enamine 12a then engages in the rate-determin-
ing B(C6F5)3-catalyzed intermolecular hydrosilylation[8b]
through the Piers mechanism[20] with 12a as a carbon
nucleophile (B(C6F5)3!13a!15aa). Alternatively,
B(C6F5)3-activated hydrosilane 13a can also react with the
amine nitrogen nucleophile 1a to equilibrate with silylam-
monium borohydride 14aa, the resting species of the overall
process.[8a,b] Initially formed 15aa stands in equilibrium with
regioisomeric 15aa
’
and 15aa
’’
, and 15aa
’’
can undergo
another deprotonation affording enamine 16aa. That enam-
ine again enters the catalytic cycle of the B(C6F5)3-promoted,
now intramolecular hydrosilylation to eventually arrive at the
title compound 3aa.
In summary, we have developed a B(C6F5)3-catalyzed two-
fold b,b’-selective (formal) C(sp3)H silylation of acyclic
tertiary amines with dihydrosilanes to construct 4-silapiper-
idines and its derivatives. The reaction involves two amine-to-
enamine dehydrogenation reactions each followed by an
inter- and an intramolecular electrophilic enamine silylation,
respectively.
Acknowledgements
H.F. gratefully acknowledges the Alexander von Humboldt
Foundation for a postdoctoral fellowship (2018–2020), and
K.X. thanks the China Scholarship Council for a predoctoral
fellowship (2019–2023). M.O. is indebted to the Einstein
Foundation Berlin for an endowed professorship. We also
thank Dr. Maria Schlangen (TU Berlin) for expert advice
with the MS measurements. Open access funding enabled and
organized by Projekt DEAL.
Conflict of interest
The authors declare no conflict of interest.
Keywords: amines · boron · CH activation · SiH activation ·
silicon
[1] C. He, W. G. Whitehurst, M. J. Gaunt, Chem 2019,5, 1031–1058,
and references therein.
[2] For authoritative reviews, see: a) S. C. Richter, M. Oestreich,
Trends Chem. 2020,2, 13–27; b) J. F. Hartwig, E. A. Romero,
Tetrahedron 2019,75, 4059– 4070; c) Y. Fukumoto, N. Chatani in
Organosilicon Chemistry: Novel Approaches and Reactions
(Eds.: T. Hiyama, M. Oestreich), Wiley-VCH, Weinheim, 2019,
pp. 171 –211.
[3] For examples of transition-metal-catalyzed C(sp3)H silylation
aor bto an amine nitrogen atom, see: a) T. Mita, K. Michigami,
Y. Sato, Chem. Asian J. 2013,8, 2970–2973 (a, directed, and
intermolecular); b) H. Fang, W. Hou, G. Liu, Z. Huang, J. Am.
Chem. Soc. 2017,139, 11601–11609 (aand intramolecular);
c) B. Su, T. Lee, J. F. Hartwig, J. Am. Chem. Soc. 2018,140,
18032– 18038 (band intramolecular).
[4] For a review, see: a) S. Park, Chin. J. Chem. 2019,37, 1057–1071;
for a review dedicated to transition-metal-free CH silylation,
see: b) D. P. Schuman, W.-B. Liu, N. Nesnas, B. M. Stoltz in
Organosilicon Chemistry: Novel Approaches and Reactions
(Eds.: T. Hiyama, M. Oestreich), Wiley-VCH, Weinheim, 2019,
pp. 213 –240.
[5] For examples of B(C6F5)3-catalyzed a-functionalization of
tertiary amines, see: a) M. Shang, J. Z. Chan, M. Cao, Y.
Chang, Q. Wang, B. Cook, S. Torker, M. Wasa, J. Am. Chem.
Soc. 2018,140, 10593–10601; b) A. F. G. Maier, S. Tussing, H.
Zhu, G. Wicker, P. Tzvetkova, U. Flçrke, C. G. Daniliuc, S.
Grimme, J. Paradies, Chem. Eur. J. 2018,24, 16287–16291; c) J.-
J. Tian, N.-N. Zeng, N. Liu, X.-S. Tu, X.-C. Wang, ACS Catal.
2019,9, 295–300; d) J. Z. Chan, Y. Chang, M. Wasa, Org. Lett.
2019,21, 984– 988; e) J. Z. Chan, A. Yesilcimen, M. Cao, Y.
Zhang, B. Zhang, M. Wasa, J. Am. Chem. Soc. 2020,142, 16493–
16505; f) S. Basak, A. Alvarez-Montoya, L. Winfrey, R. L.
Melen, L. C. Morrill, A. P. Pulis, ACS Catal. 2020,10, 4835–
4840.
[6] For a review, see: a) F. Focante, P. Mercandelli, A. Sironi, L.
Resconi, Coord. Chem. Rev. 2006,250, 170–188; for a key
publication, see: b) N. Millot, C. C. Santini, B. Fenet, J. M.
Basset, Eur. J. Inorg. Chem. 2002, 3328–3335.
[7] These ammonium borohydrides are dihydrogen adducts of
amine/borane FLPs (FLP=frustrated Lewis pair). While release
of dihydrogen is slow at room temperature, reversible dihydro-
gen activation occurs at elevated temperatures. V. Sumerin, F.
Schulz, M. Nieger, M. Leskel, T. Repo, B. Rieger, Angew.
Chem. Int. Ed. 2008,47, 6001–6003; Angew. Chem. 2008,120,
6090–6092.
[8] a) J. Zhang, S. Park, S. Chang, J. Am. Chem. Soc. 2018,140,
13209– 13213; see also: b) M. Zhou, S. Park, L. Dang, Org.
Chem. Front. 2020,7, 944–952; c) J. Zhang, S. Chang, J. Am.
Chem. Soc. 2020,142, 12585–12590.
[9] R. Li, Y. Chen, K. Jiang, F. Wang, C. Lu, J. Nie, Z. Chen, G. Yang,
Y.-C. Chen, Y. Zhao, C. Ma, Chem. Commun. 2019,55, 1217–
1220.
Scheme 6. Plausible mechanism for the formation of 3aa from 1a and
2a (Si=HPh2Si). rds=rate-determining step.
A
ngewandte
Chemi
e
Communications
8545Angew. Chem. Int. Ed. 2021,60, 8542 –8546 2021 The Authors. Angewandte Chemie International Edition published by Wiley-VCH GmbH www.angewandte.org

[10] Y. Chang, A. Yesilcimen, M. Cao, Y. Zhang, B. Zhang, J. Z.
Chan, M. Wasa, J. Am. Chem. Soc. 2019,141, 14570–14575.
[11] Y. Chen, H.-L. Wan, Y. Huang, S. Liu, F. Wang, C. Lu, J. Nie, Z.
Chen, G. Yang, C. Ma, Org. Lett. 2020,22, 7797–7803.
[12] a) Y. Ma, B. Wang, L. Zhang, Z. Hou, J. Am. Chem. Soc. 2016,
138, 3663– 3666; b) Q. Yin, H. F. T. Klare, M. Oestreich, Angew.
Chem. Int. Ed. 2016,55, 3204–3207; Angew. Chem. 2016,128,
3256–3260; for a review, see: c) S. Bhr, M. Oestreich, Angew.
Chem. Int. Ed. 2017,56, 52–59; Angew. Chem. 2017,129, 52– 59.
[13] For a review, see: a) A. K. Franz, S. O. Wilson, J. Med. Chem.
2013,56, 388– 405; for recent work on silicon-containing nitro-
gen heterocycles, see: b) S. J. Barraza, S. E. Denmark, J. Am.
Chem. Soc. 2018,140, 6668–6684.
[14] The 4-silapiperidine core was prepared in several steps with
a ring-forming dialkylation of a primary amine as the key step
(90
8
C in an autoclave for 16 h): R. Tacke, T. Heinrich, R.
Bertermann, C. Burschka, A. Hamacher, M. U. Kassack, Orga-
nometallics 2004,23, 4468–4477.
[15] a) M. Gerlach, P. Jutzi, J.-P. Stasch, H. Przuntek, Z. Naturforsch.
B1982,37, 657–662; b) B. M. Kim, J. H. Cho, Tetrahedron Lett.
1999,40, 5333–5336; for hydroamination with lithium amides,
see: c) Ref. [15b]; for formal hydroamination by aminomercu-
ration–reduction, see: d) J. Barluenga, C. Jimnez, C. Njera, M.
Yus, Synthesis 1982, 414 – 417.
[16] The roles of these additives are not entirely clear. The metal
oxides are believed to facilitate the deprotonation step[8a]
(iminium ion!enamine; Scheme 1, gray box) while silyl triflates
are thought to enhance hydride release from the borohydride
through the intermediate formation of a pentacoordinate silicon
hydride as a hydride shuttle.[5c]
[17] a) H. Fang, M. Oestreich, Angew. Chem. Int. Ed. 2020,59,
11394– 11398; Angew. Chem. 2020,132, 11491–11495; see also:
b) Ref. [8c].
[18] Such spirocyclic systems are also relevant in medicinal chemis-
try: R. Tacke, V. I. Handmann, R. Bertermann, C. Burschka, M.
Penka, C. Seyfried, Organometallics 2003,22, 916– 924.
[19] a) A. F. G. Maier, S. Tussing, T. Schneider, U. Flçrke, Z.-W. Qu,
S. Grimme, J. Paradies, Angew. Chem. Int. Ed. 2016,55, 12219–
12223; Angew. Chem. 2016,128, 12407–12411; b) M. Kojima, M.
Kanai, Angew. Chem. Int. Ed. 2016,55, 12224– 12227; Angew.
Chem. 2016,128, 12412– 12415.
[20] a) D. J. Parks, J. M. Blackwell, W. E. Piers, J. Org. Chem. 2000,
65, 3090– 3098; b) S. Rendler, M. Oestreich, Angew. Chem. Int.
Ed. 2008,47, 5997– 6000; Angew. Chem. 2008,120, 6086–6089;
c) K. Sakata, H. Fujimoto, J. Org. Chem. 2013,78, 12505–12512;
d) A. Y. Houghton, J. Hurmalainen, A. Mansikkamki, W. E.
Piers, H. M. Tuononen, Nat. Chem. 2014,6, 983–988.
Manuscript received: December 15, 2020
Revised manuscript received: February 16, 2021
Accepted manuscript online: February 19, 2021
Version of record online: March 3, 2021
A
ngewandte
Chemi
e
Communications
8546 www.angewandte.org 2021 The Authors. Angewandte Chemie International Edition published by Wiley-VCH GmbH Angew. Chem. Int. Ed. 2021,60, 8542 –8546
Loading more pages...