
ARTICLE
Morphology and mechanism of highly selective
Cu(II) oxide nanosheet catalysts for carbon dioxide
electroreduction
Xingli Wang 1, Katharina Klingan 2, Malte Klingenhof1, Tim Möller 1, Jorge Ferreira de Araújo1,
Isaac Martens3, Alexander Bagger 4, Shan Jiang2, Jan Rossmeisl 4, Holger Dau 2& Peter Strasser 1✉
Cu oxides catalyze the electrochemical carbon dioxide reduction reaction (CO2RR) to
hydrocarbons and oxygenates with favorable selectivity. Among them, the shape-controlled
Cu oxide cubes have been most widely studied. In contrast, we report on novel 2-dimensional
(2D) Cu(II) oxide nanosheet (CuO NS) catalysts with high C
2+
products, selectivities
(> 400 mA cm−2) in gas diffusion electrodes (GDE) at industrially relevant currents and
neutral pH. Under applied bias, the (001)-orientated CuO NS slowly evolve into highly
branched, metallic Cu0dendrites that appear as a general dominant morphology under
electrolyte flow conditions, as attested by operando X-ray absorption spectroscopy and
in situ electrochemical transmission electron microscopy (TEM). Millisecond-resolved dif-
ferential electrochemical mass spectrometry (DEMS) track a previously unavailable set of
product onset potentials. While the close mechanistic relation between CO and C
2
H
4
was
thereby confirmed, the DEMS data help uncover an unexpected mechanistic link between
CH
4
and ethanol. We demonstrate evidence that adsorbed methyl species, *CH
3
, serve as
common intermediates of both CH
3
H and CH
3
CH
2
OH and possibly of other CH
3
-R products
via a previously overlooked pathway at (110) steps adjacent to (100) terraces at larger
overpotentials. Our mechanistic conclusions challenge and refine our current mechanistic
understanding of the CO
2
electrolysis on Cu catalysts.
https://doi.org/10.1038/s41467-021-20961-7 OPEN
1Department of Chemistry, Chemical Engineering Division, Technical University Berlin, Straße des 17. June 124, 10623 Berlin, Germany. 2Department of
Physics, Free University of Berlin, Arnimallee 14, 14195 Berlin, Germany. 3European Synchrotron Radiation Facility (ESRF), 38000 Grenoble, France.
4Department of Chemistry, University of Copenhagen, Copenhagen 2100, Denmark. ✉email: [email protected]
NATURE COMMUNICATIONS | (2021) 12:794 | https://doi.org/10.1038/s41467-021-20961-7 |www.nature.com/naturecommunications 1
1234567890():,;

Valorizing atmospheric CO
2
into C
2+
products by electro-
chemical method holds the promise to store renewable
surplus electricity while closing the global carbon cycle1–3.
Efficient CO
2
electrolysis requires advanced electrocatalyst
designs4–10, an understanding of competing reaction
pathways11,12, and a reliable device performance at industrial
conditions13–16. Among a wide variety of catalysts tested for
electrochemical CO
2
reduction reaction (CO2RR), copper-based
materials, in particular Cu oxides, CuO
x
, have been enjoying
attention thanks to their wide chemical selectivity for a variety of
multi-carbon products, such as C
2+
hydrocarbons and oxyge-
nates. It has been reported that over 16 kinds17 of products can be
formed on copper catalysts through multiple proton-coupled
electron transfer processes.
The cathodic electrode potentials during the catalytic CO2RR
invariably drive the chemical transformation of operating CuO
x
catalysts into metallic Cu phases. This prompted their designation
as oxide-derived copper (OD-Cu) catalysts. As a consequence of
the chemical reduction metallic Cu0, complex time trajectories of
catalyst morphology, chemical state, and catalytic selectivity
ensues. A full molecular correlation and understanding of these
concomitant trajectories has remained elusive. It has been sug-
gested that the increased local pH value18–20 of OD-Cu, in direct
proportion to the surface roughness, plays an important role for
favorable ethylene selectivity compared to methane21. Other
works highlighted the distinct chemisorption of CO inter-
mediates22–24. Yet other reports related the catalytic performance
to grain boundaries25, to undercoordinated sites26, to the pre-
sence of subsurface oxygen27,28 or to residual near-surface Cu+29.
Also, in most studies, the end point of the morphological evo-
lution of designer OD-Cu catalysts has remained in the dark.
Shape-selected cubic Cu
2
Ocatalysts—largely obtained
through potential cycling in presence of surface-active electrolyte
additives—have been receiving particular attention as active and
selective CO2RR electrocatalysts30,31. Thanks to the structure
sensitivity of the C-C bond formation on square atomic Cu0
surface lattices and the cubic nature of the Cu
2
O crystal system,
the resulting preferential Cu(100) facets offer kinetic faradaic
efficiency and onset potential benefits for the formation of C
2+
products, in particular C
2
H
4
32.
To follow the evolution in chemical state and morphology of
electrocatalysts under stationary electrode potential conditions,
operando X-ray-based33 as well as in situ vibrational
spectroscopies31,34–36, and in situ electrochemical transmission
electron microscopy (TEM)37–39 were used to record changes in
catalyst chemical state40,41, local structure13, or intermediates42.
However, to date, real-time tracking individual CO2RR product
yields and product onset potentials on OD-Cu catalysts at fast
time scales under non-stationary, transient conditions has
remained very challenging. This operational mode, however, is
very relevant for practical CO2RR electrolyzers powered by
intermittent input electricity from renewable sources.
Even though Cu-based CO2RR electrocatalysts have shown
excellent C
2
H
4
efficiencies in highly alkaline pH 13–15
conditions13,43, practical CO2RR electrocatalysis must operate at
neutral pH ~ 7 conditions and perform at industrially relevant
current densities > 200 mA cm−213,14. To achieve this, the cata-
lysts must be cast in gas diffusion electrodes (GDEs) that are
deployed in single zero- and non-zero gap flow electrolyzer cells.
While a few recent studies have reported the use of free-standing
cubic Cu
2
O nanocubes44–46 inside GDE designs, the majority of
OD-Cu based GDE studies relied on top-down approaches
involving modified bulk Cu43. Compared to bulk Cu-based cat-
alysts, nanostructured free-standing catalysts are more desirable
as they exhibit higher surface-to-volume ratio and are more
amenable to assembly in large-scale GDEs.
In this contribution, we report on a new family of free-standing
2-dimensional Cu(II) oxide electrocatalysts for the CO2RR under
neutral pH conditions. Owing to their 2D nature, Cu(II)O
nanosheets (referred to as “2D CuO NS”) feature a highly pre-
ferred (001) facet orientation. We report a facile one-step
synthesis of 2D CuO NS and then track their unique catalytic
reactivity and concomitant morphological evolution under
applied electrode potentials using in situ electrochemical liquid
TEM and operando X-ray absorption spectroscopy (XAS). Next,
using a new differential electrochemical mass spectrometry
(DEMS) technique with millisecond resolution, we discuss pre-
viously unreported time evolutions of kinetic onset potentials of a
range of different products over the course of hours. Conclusions
from our DEMS data challenge and refine our current mechan-
istic understanding of the mechanistic link between CH
4
and
ethanol (EtOH). Finally, we document the favorable CO2RR
performance of 2D CuO NS inside GDEs of commercial elec-
trolyzers under industrially relevant neutral pH conditions.
Owing to their emerging stable dendritic structure, their high
catalytic reactivity, and the resulting performance stability,
oriented 2D CuO NS precursor catalysts constitute an interesting
alternative to conventional cubic Cu
2
O catalysts for the electro-
chemical conversion of CO
2
into C
2
H
4
.
Results
Synthesis and characterization of 2-dimensional CuO
nanosheets. Free-standing CuO nanosheet catalysts (CuO NS)
were prepared by solvothermal formation and processing of Cu
(OH)
2
intermediates in alkaline condition (Supplementary
Fig. S2). The wrinkled thin Cu(OH)
2
layers showed high aspect
ratios prior to decomposing into CuO and H
2
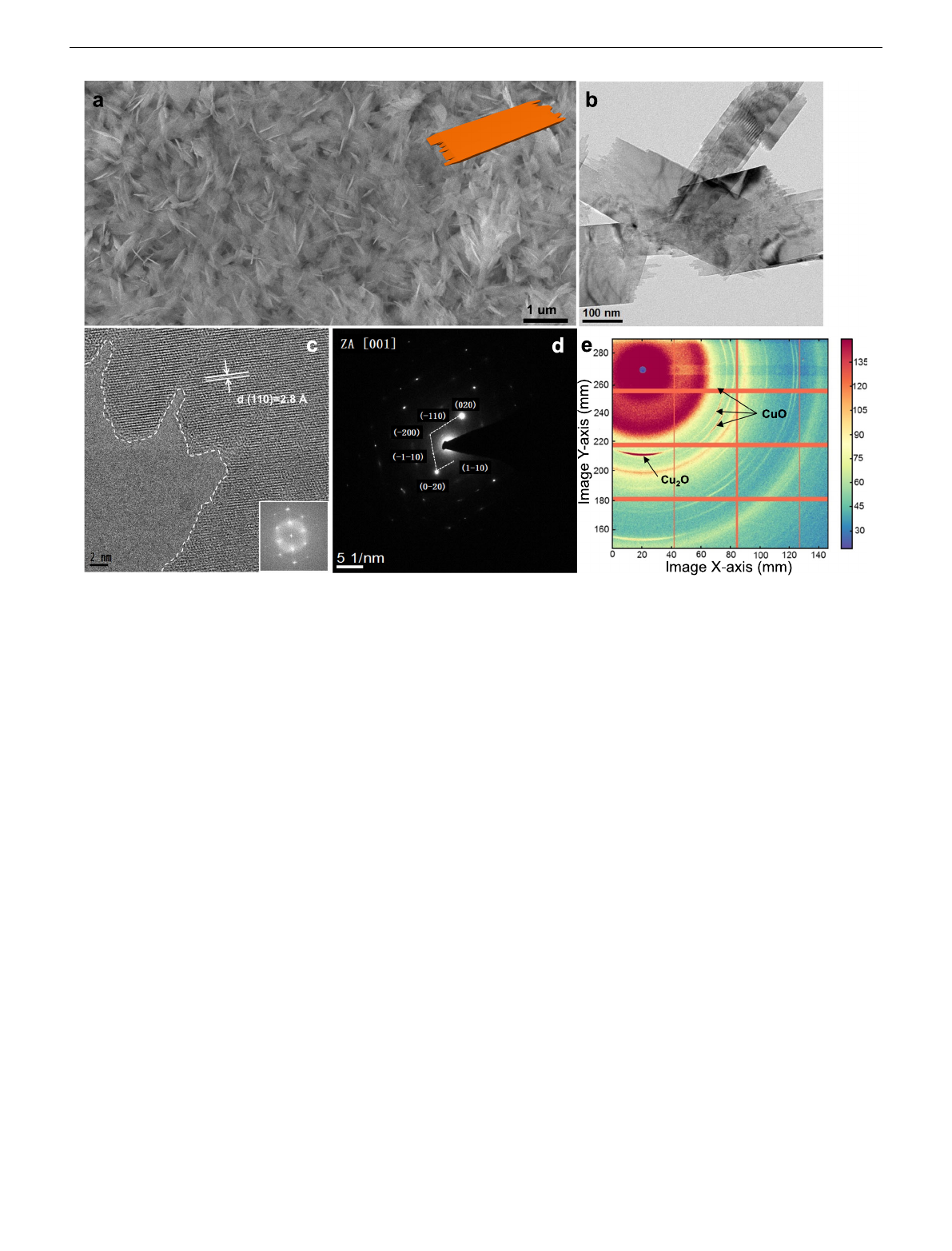
O. Figure 1a,b
presents scanning electron microscopy (SEM) image and trans-
mission electron microscopy (TEM) image of as-prepared CuO
NS. The 2D character of the material was retained during the
thermal decomposition of Cu(OH)
2
, while larger sheets split into
small ones. The final rectangular CuO NS exhibited serrated
edges on the short edges. Supplementary Fig. S3a indicates a
stacked structure of individual free-standing CuO NS of 3−4nm
thickness. High-resolution transmission electron microscopy
(HR-TEM) (Fig. 1c) of CuO NS revealed well-defined lattice
fringes with an interplanar spacing of 2.8 Å, corresponding to
{110} planes of monoclinic CuO. Selected area electron diffraction
(SAED) images (Fig. 1d) showed rhombic diffraction spots along
the [001] zone axis. This can be seen by indexed (020), (-110),
(-200), (-1-10), (0-20) and (1-1;0) planes, indicating that the CuO
NS had {001} exposed surfaces. The relative intensity of the dif-
fraction spots is presented in Supplementary Fig. S3b. The crystal
structure is provided in Supplementary Fig. S4 with indexed
(001), (110) and (11-1) planes.
A 2D grazing-incidence wide-angle X-ray scattering (GI-WAXS)
pattern of CuO NS on glassy carbon electrode is shown in Fig. 1e.
The pattern is commensurate with a crystalline CuO phase. Minor
contributions of a residual Cu
2
O phase are seen at low angles,
yet all higher order reflections were absent evidencing their trace
character. Importantly, the (002) and (11-1) reflections of CuO
exhibited a stronger intensity in the meridional direction,
evidencing that the CuO NS are stacked along the <00 l> direction,
supporting the HR-TEM results. A partial contour plot and
integrated CuO NS line profiles are shown in Supplementary
Fig. S5, indicating the coexistence of Cu
2
O traces and major
CuO phase.
CO2RR reactivity, stability, and ex situ morphology in H-cells.
The catalytic CO2RR activity of the CuO NS in pH neutral
conditions and without any further pretreatment was recorded
ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-021-20961-7
2NATURE COMMUNICATIONS | (2021) 12:794 | https://doi.org/10.1038/s41467-021-20961-7 | www.nature.com/naturecommunications
for up to 60 h. Product yields as function of iR-corrected electrode
potential (E
RHE
) after 1 h electrolysis are plotted in Fig. 2a. C
2
H
4
started to evolve at significantly more anodic electrode potentials
compared to CH
4
, which offered a roughly 300 mV-wide poten-
tial window of exclusive C
2
H
4
production (their detailed time-
resolved onset potentials see DEMS section below). CH
4
forma-
tion showed a rapid growth beyond −0.9 V
RHE
and exceeded
C
2
H
4
production at −0.97 V
RHE
. The observed partial current
densities under the chosen conditions (Fig. 2b) are testament to
previously unachieved hydrocarbons selectivities under pH neu-
tral conditions. The partial current density of C
2
H
4
reached
6.2 mA cm−2at −0.97 V
RHE
, while CO remained at very low
levels during the entire given overpotential range. The Faradaic
efficiencies (FEs) within the potential window of exclusive C
2
H
4
production and beyond are compared in Supplementary Fig. S6.
The suppression of the HER and concomitant increase in C
2
H
4
are noteworthy for H-cell studies.
Longer-term 20 h and 60 h electrolysis was carried out to
track the CuO NS selectivity and stability at selected potentials
(Fig. 2c−e). CuO NS catalysts maintained a stable absolute C
2
H
4
production rate during the 60 h electrolysis, even past
replacements of the electrolyte (Fig. 2d, e). By contrast, the
CH
4
production decreased and dropped to very low values after
25 h, making CuO NS voltage efficient (low overpotentials),
C
2
H
4
-selective and very performance-stable electrocatalysts.
The initial catalyst activation period over the first few hundred
minutes merits a closer time resolution of the reactivity coupled to a
correlation to the sheet morphology (Supplementary Figs. S7−S10).
The time trajectories of CO, CH
4
,andC
2
H
4
production
(Supplementary Fig. S7) exhibited similar patterns over the first
2−3 h of CuO NS catalyst activation. The hydrocarbon production
rate peaked earlier with increasing applied overpotential. Consistent
with data from Fig. 2a, exclusive and sustained catalytic C
2
H
4
production was detected at −0.84 V
RHE
at 1.6 nmol cm−2s−1,
exceeding previous reports47.Near−1.0 V
RHE
, hydrocarbon
production peaked already after 2 h, followed by a steady drop in
CH
4
generation (cf. Fig. 2d).
Morphological changes of CuO NS (Supplementary Fig. S8)
after the first hour electrolysis near −1.0 V
RHE
involved rounding
of the nanosheet with agglomerations. After prolonged electro-
lysis at −0.76 V
RHE
and −0.84 V
RHE
(Supplementary Figs. S9,
S10), the rounding led to a sheet fragmentation of the initial CuO
NS appeared to be more obvious. Finally, after a 60-h electrolysis
at −1.0 V
RHE
, the sheet fragments re-assembled into larger
agglomerates with rough surfaces (Supplementary Fig. S11). In a
comparative study of the evolution of the catalyst morphology as
a function of electrode potential under identical conditions, the
CuO NS catalyst was drop cast on a rough carbon fiber paper
rather than on smooth glassy carbon. Thanks to better dispersed
NS, the catalytic current density increased (Supplementary
Fig. S12). Now, ex situ SEM studies after 1 h reaction
(Supplementary Fig. S13) evidenced that rough supports slowed
down the morphological transformations of the CuO NS. Unlike
shown in Supplementary Fig. S8, part of the sheet morphology
appears still intact (Supplementary Fig. S13c, Area 3), with
individual sheets on the fiber cracked (Supplementary Fig. S13b,
Area 1) and fractured into small clusters (Supplementary
Fig. S13b, Area 2).
Fig. 1 Morphological, structural characterizations of sheet-like CuO catalysts synthesized in this study by ex situ techniques. a Large-scale scanning
electron microscopy (SEM) image and 3D structure of CuO NS (insert, orange). bTransmission electron microscopy (TEM) image and chigh resolution
TEM (HR-TEM) images with measured lattice distance and the corresponding fast Fourier transformation. dSelected area electron diffraction (SAED)
pattern along zone axis [001]. e2D Synchrotron grazing incidence wide-angle X-ray scattering (GI-WAXS) image demonstrating preferred orientation of
as-prepared CuO NS on glassy carbon. Additional azimuthally integrated line profiles are shown in Supplementary Fig. S5b.
NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-021-20961-7 ARTICLE
NATURE COMMUNICATIONS | (2021) 12:794 | https://doi.org/10.1038/s41467-021-20961-7 |www.nature.com/naturecommunications 3
Real-time morphology using in situ electrochemical TEM—
from sheets to dendrites. The ex situ microscopic studies
revealed rapid (< 1 h) fragmentation of CuO NS during CO2RR
associated with rapid kinetic activation and monotonically rising
product selectivity (Supplementary Figs. S7, S8 and S13). Of
particular interest was the applied bias of −0.84 V
RHE
, where
hydrocarbon generation was limited exclusively to C
2
H
4
. To learn
more about the NS catalyst morphology that enabled exclusive
C
2
H
4
-selectivity, we followed the transformations of the CuO NS
at that same bias using in situ electrochemical liquid TEM
experiments. These studies were carried out in a Protochips
Poseidon holder that hosted a flow-through electrochemical chip
cell (“in situ TEM E-chip cell”, Supplementary Fig. S14b)
equipped with parallel beam-transparent silicon nitride windows.
The top view of the in situ TEM E-chip cell is illustrated in
Supplementary Fig. S14a. To exclude beam damage and liquid
layer effects, all in situ TEM E-chip cell experiments were cross-
checked using identical location (IL) TEM E-chip cells, in which
the E-chip cell was operated outside the microscope under
otherwise identical conditions (Supplementary Fig. S14c), yet
imaged in the dry state. The in situ studies cover the first few
minutes of the catalyst activation where the most dramatic rise in
catalytic activity occurred (Supplementary Fig. S7).
First, Supplementary Movie 1 tracks the initial morphological
evolution of the CuO NS under open circuit potential (OCP) and
pH-neutral aqueous conditions. Three selected times (Supple-
mentary Fig. S15) and their TEM snapshots are shown in
Fig. 3a–c. CuO NS of several 100 s of nm in size fracture into
smaller sheet fragments, however without immediate disintegra-
tion. Over the 110 s imaging time, some of the fragmented sheets
slowly drifted out of view into the liquid layer.
To check the influence of beam and liquid layer effects, we
conducted a prolonged 40 min OCP experiment in the IL TEM E-
chip cell. In agreement with the in situ results, the CuO NS with
100 s of nanometer size fractured into fragments of few nanometer
size (Supplementary Fig. S16b, c) without disintegration.
Next, we used the in situ TEM E-chip cell to conducted real-
time imaging of the CuO NS in the pH =6.9 buffer solution, still
-1.00 -0.95 -0.90 -0.85 -0.80 -0.75
0.01
0.1
1
10
100
E
RHE
Production Rate
(nmol cm
-2
s
-1
)
(nmol cm
-2
s
-1
)
H2
CO
CH4
C2H4
1h
-1.00 -0.95 -0.90 -0.85 -0.80 -0.75
0
2
4
6
8
10
Partial Current Density
E
RHE
(V)
1h
H2
CO
CH4
C2H4
02468101214161820
0
3
6
9
12
15
18
CH
4
CO
C
2
H
4
Production Rate
Time
H
2
@-0.84 V
RHE
Solid
0
10
20
30
40
50
FE
Hollow
0 10205060
0
10
20
30
40
50
H2
CO
CH4
C2H4
Time
Production Rate
New Electrolyte
@ -1.0 V
RHE
0 10205060
0
10
20
30
40
50
H
2
CO
CH
4
C
2
H
4
Time
FE
New Electrolyte
@ -1.0 VRHE
ab
d
c
e
(V)
mcAm( 2
-)
(nmol cm
-2
s
-1
)
(%)
(h)
(h) (h)
(%)
Fig. 2 Electrocatalytic CO2RR tests using CuO NS in H-cells. a Absolute product formation rates of major gaseous products as a function of applied
electrode potentials during CO2RR in CO
2
-saturated 0.1 M KHCO
3
at 60 min. bPartial current densities as a function of applied electrode potentials during
CO2RR in CO
2
-saturated 0.1 M KHCO
3
at 60 min. cChronoamperometric performance stability of the CO
2
reduction reaction on CuO NS in CO
2
-saturated
0.1 M KHCO
3
at −0.84 V
RHE
.dand eLong-term stability test over 60 h for dabsolute product formation rates of major gaseous products and eFaradaic
efficiencies on CuO NS in CO
2
-saturated 0.1 M KHCO
3
at −1.0 V
RHE
. The error bars are given as standard error of mean. Catalyst loading: 100 μgcm
−2.
ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-021-20961-7
4NATURE COMMUNICATIONS | (2021) 12:794 | https://doi.org/10.1038/s41467-021-20961-7 | www.nature.com/naturecommunications
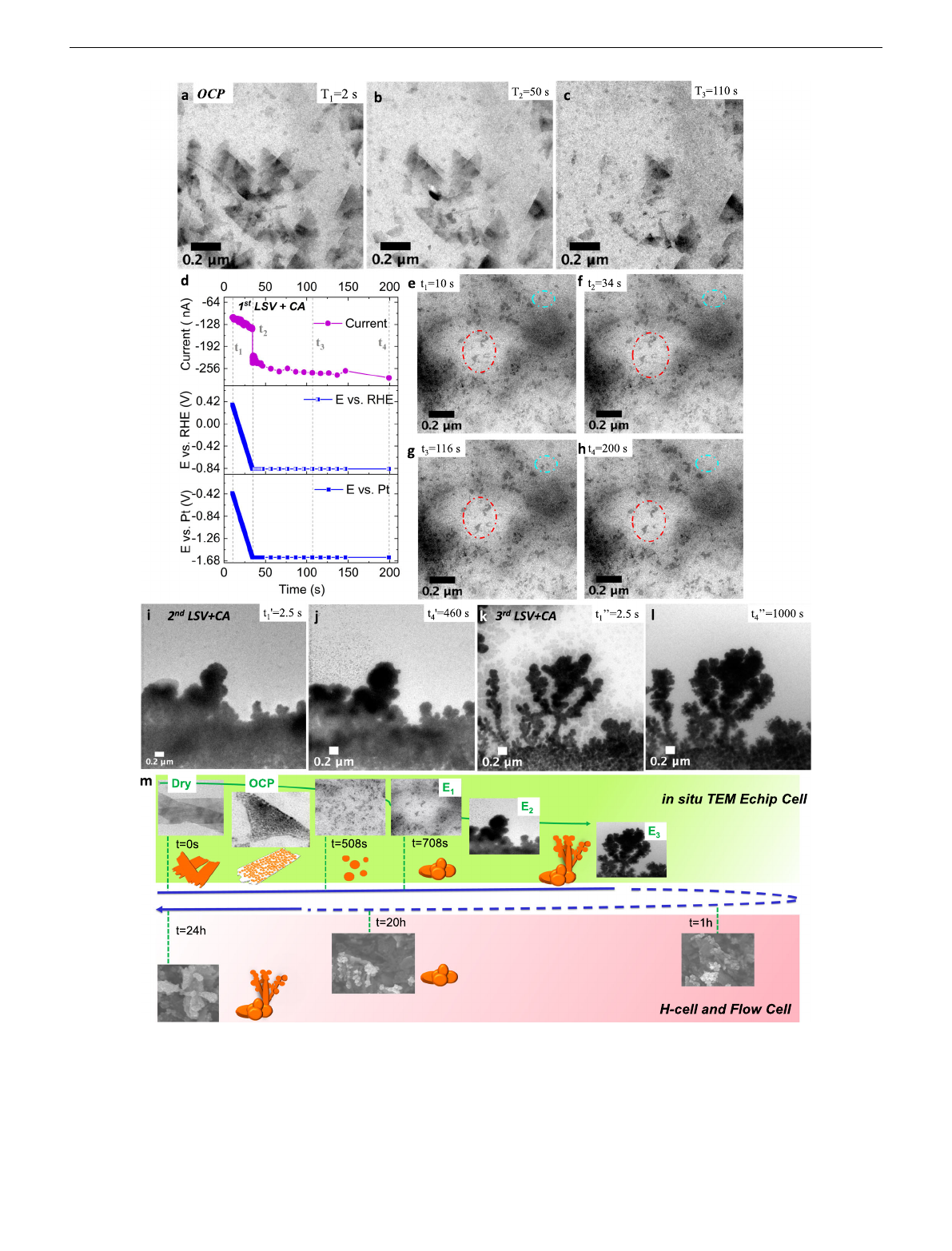
maintaining OCP. Supplementary Movies 2 and 3 captured the
structural transformations over the next ~ 400 s: Supplementary
Movie 2 documents the continued fragmentation of the CuO NS
in real time at OCP over 200 s. Clouds of dark spherical Cu
fragments are distributed non-uniformly across the field of view,
while floating and drifting in and out of focus. Over the next
198 s, Supplementary Movie 3 captures the dynamics of the
fragmented CuO NS catalyst. We believe that these sheet
fragments served as building blocks for later re-agglomeration.
Next, we investigated the effect of applied electrode bias of −0.84
V
RHE
on the subsequent morphological evolution of the
fragmented Cu catalyst. After 10 s OCP (Supplementary
Fig. S17a), a linear sweep voltammetry (LSV) step lowered the
electrode potential from +0.42 V
RHE
to −0.84 V
RHE
, where
chronoamperometric (CA) control kept it for another 170 s.
Figure3d shows the experimental current/potential profiles
NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-021-20961-7 ARTICLE
NATURE COMMUNICATIONS | (2021) 12:794 | https://doi.org/10.1038/s41467-021-20961-7 |www.nature.com/naturecommunications 5
Loading more pages...