Synthesis of Silylated Cyclobutanone and Cyclobutene
Derivatives Involving 1,4-Addition of Zinc-Based Silicon
Nucleophiles
Ming Cui[a] and Martin Oestreich*[a]
Abstract: A copper-catalyzed conjugate silylation of various
cyclobutenone derivatives with Me2PhSiZnCl·2LiCl or
(Me2PhSi)2Zn·xLiCl (x�4) to generate β-silylated cyclobuta-
nones is reported. Trapping the intermediate enolate with
ClP(O)(OPh)2affords silylated enol phosphates that can be
further engaged in Kumada cross-coupling reactions to
yield silylated cyclobutene derivatives.
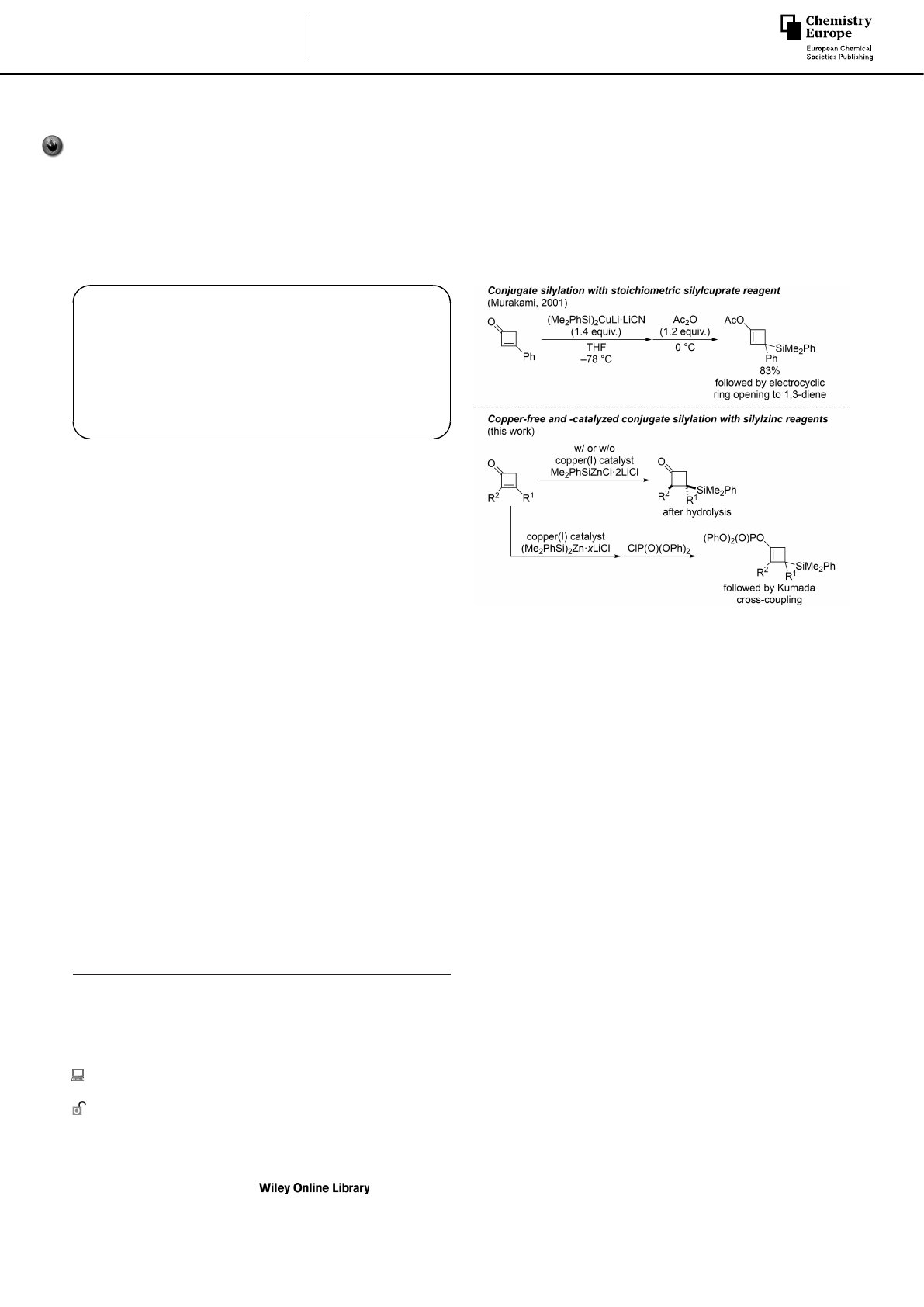
Conjugate addition of silicon nucleophiles to α,β-unsaturated
carbonyl compounds is one of the standard processes for the
formation of C(sp3)Si bonds.[1] The resulting β-silylated
carbonyl compounds[2] can be converted into the correspond-
ing aldols by oxidative degradation of that C(sp3)Si bond.[3] As
to cyclic acceptors, the vast majority of protocols are for
cyclopentenone and -hexenone derivatives.[4,5] Murakami and
co-workers reported the 1,4-addition to cyclobutenone deriva-
tives using Fleming’s (Me2PhSi)2CuLi·LiCN[4a,b] to access func-
tionalized 1,3-dienes after trapping of the enolate intermediate
and electrocyclic ring-opening (Scheme 1, top).[6] Aside from
this isolated example, there are no further methods known,
neither stoichiometric nor catalytic in copper.
Almost 20 years ago, our laboratory introduced copper-
catalyzed and even copper-free protocols for conjugate
silylation employing bis(triorganosilyl)zinc and tris(triorgano-
silyl)zincate reagents.[7–9] We also found copper salts to accel-
erate these reactions and to be essential for hindered and β,β-
disubstituted acceptors, respectively.[8] Zinc-based silicon nucle-
ophiles such as (Me2PhSi)2Zn·4LiCl and also Me2PhSiZnCl·2LiCl
are in fact highly useful. Their functional-group tolerance is
substantially improved over that of the corresponding more
reactive lithium compounds from which the zinc reagents are
typically prepared by transmetalation. To date, none of these
protocols have been applied to cyclobutenones. Moreover, the
synthesis of cyclobutyl-substituted silanes is limited to a few
examples. In 2010, Ito and co-workers reported a copper-
catalyzed borylation of silyl-substituted homoallylic sulfonates,
and cyclobutylsilane derivatives were obtained by insertion of
the CC double bond into an in situ formed CuB bond
followed by an intramolecular SN2 reaction.[10] The Fu group[11]
and our group[12] reported single examples of the synthesis of
cyclobutylsilanes by metal-catalyzed radical cross-coupling of a
tertiary and a secondary cyclobutyl bromide with zinc- and
magnesium-based silicon reagents, respectively. In this work,
we describe copper-catalyzed conjugate silylations of highly
substituted cyclobutenone derivatives with zinc-based silicon
reagents (Scheme 1, bottom). The intermediate metal enolates
can either be hydrolyzed to afford 3-silyl-substituted cyclo-
butanones or captured with ClP(O)(OPh)2as an electrophile to
furnish cyclobutenyl phosphates. Subsequent Kumada cross-
coupling yields silicon-containing cyclobutene derivatives.
Our study commenced with the conjugate silylation of
cyclobutenone 1a with 2.0 equiv. of Me2PhSiZnCl·2LiCl in
THF[13] (Table 1). Using Cu(CH3CN)4PF6as the catalyst in THF at
room temperature, β-silylated β-phenylcyclobutanone 2a was
obtained in 95% yield after hydrolysis (entry 1). Yields were
slightly lower with less silicon nucleophile, for example 91%
yield with 1.5 equiv. of Me2PhSiZnCl·2LiCl. Given the possibility
of a copper-free 1,4-addition,[8] we compared different β-
[a] M. Cui, Prof. Dr. M. Oestreich
Institut für Chemie
Technische Universität Berlin
Strasse des 17. Juni 115, 10623 Berlin (Germany)
E-mail: [email protected]
Supporting information for this article is available on the WWW under
https://doi.org/10.1002/chem.202102993
© 2021 The Authors. Chemistry - A European Journal published by Wiley-
VCH GmbH. This is an open access article under the terms of the Creative
Commons Attribution License, which permits use, distribution and re-
production in any medium, provided the original work is properly cited.
Scheme 1. Conjugate silylation of cyclobutenone derivatives and follow-up
chemistry of the in situ-formed enolates.
Chemistry—A European Journal
Communication
doi.org/10.1002/chem.202102993
16103Chem. Eur. J. 2021,27, 16103–16106 © 2021 The Authors. Chemistry - A European Journal published by Wiley-VCH GmbH
Wiley VCH Donnerstag, 11.11.2021
2165 / 221299 [S. 16103/16106] 1
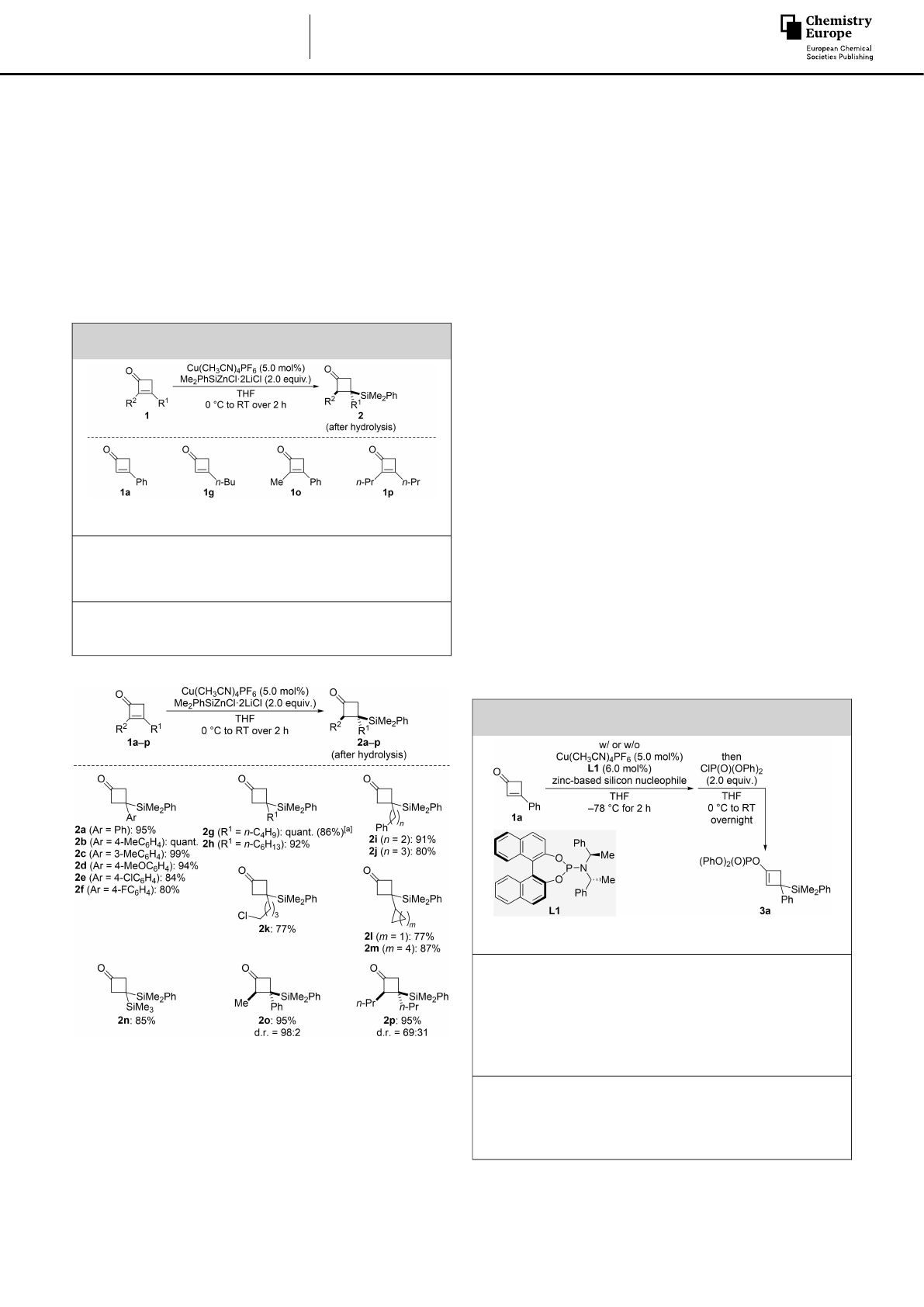
substituted and α,β-disubstituted cyclobutenones in reactions
with and without the copper catalyst. The silylation of 1a in the
absence of Cu(CH3CN)4PF6did lead to 2a yet with a substantial
decrease in yield (entry 1). Other cyclobutenones such as β-
butyl-substituted 1g and α,β-disubstituted 1o and 1p were
tested, and the low yields of the copper-free protocol confirmed
the importance of a copper catalyst (entries 2–4).
We further tested the substrate scope of this conjugate
silylation (Scheme 2). β-Aryl-substituted cyclobutenones were
generally suitable substrates, affording the corresponding β-
silylated cyclobutanones in good to excellent yields (1a–f!2a–
f). Electron-donating groups at the aryl ring such as methyl and
methoxy led to higher yields than halogenated derivatives.
Likewise, cyclobutenones bearing a primary alkyl substituent in
the β-position furnished the corresponding products in equally
high yields (1g–k!2g–k); the yield was lowest for 1 k
containing a C(sp3)Cl bond. With sterically more demanding
secondary alkyl groups such as cyclopropyl and cyclohexyl,
yields were still good (1l,m!2l,m). A silyl group in the β-
position was also compatible (1n!2n). The reactions of α,β-
disubstituted cyclobutenones 1o and 1p proceeded equally
well. Product 2o was obtained with high diastereoselectivity
while 2p formed with a poor diastereomeric ratio. We believe
that the diastereoselectivity is mainly controlled by steric factors
in the protolysis of the enolate intermediate.
Next, we tried to capture the enolate intermediate as an
enol phosphate,[14] that is cyclobutenyl phosphates 3, to allow
for subsequent cross-coupling reactions.[15] The brief survey
outlined in Table 2 shows that copper-catalyzed 1,4-addition of
either Me2PhSiZnCl· 2LiCl or (Me2PhSi)2Zn·xLiCl (x�4) to 1 a
followed by enolate trapping with ClP(O)(OPh)2furnishes the
enol phosphate 3a in moderate yields (entries 1 and 2).
Relevant to an enantioselective variant, no uncatalyzed back-
ground reaction was seen with an almost salt-free stock
solution of (Me2PhSi)2Zn· xLiCl in Et2O[16] (entry 2). In the light of
our recent work about an enantioselective conjugate silylation
with a zinc-based silicon nucleophile,[17] we decided to inves-
tigate the asymmetric version. The yield increased in the
presence of the chiral phosphoramidite ligand (S,R,R)-L1 but
enantioinduction was low, even at 78 °C (entries 3 and 4). A
Table 1. Comparison of copper-catalyzed and copper-free protocols with
Me2PhSiZnCl·2LiCl.[a]
Entry Acceptor Product Yield of 2[%][b]
w/ Cu(CH3CN)4PF6
Yield of 2[%][c]
w/ o Cu(CH3CN)4PF6
11a 2a 95 71
21g 2g quant. 24
31o 2o 95 30
41p 2p 95 0
[a] All reactions were performed on a 0.2 mmol scale for 2 h. [b] Isolated
yield after flash chromatography on silica gel. [c] Determined by 1H NMR
spectroscopy by using CH2Br2as the internal standard.
Scheme 2. Synthesis of β-silylated cyclobutanones by conjugate addition of
Me2PhSiZnCl·2LiCl. Unless otherwise noted, all reactions were performed on
a 0.2 mmol scale for 2 h. Yields are of analytically pure product obtained
after flash chromatography on silica gel. The relative configuration was
assigned by 1H NMR spectroscopic analysis prior to purification (see the
Supporting Information for details). [a] Value in parentheses for the reaction
on a 1.0 mmol scale.
Table 2. Comparison of copper-catalyzed and copper-free protocols with
enolate trapping.[a]
Entry Zinc-based silicon
nucleophile
Yield of 3a [%][b]
w/ Cu(CH3CN)4PF6
Yield of 3a [%][b]
w/ o Cu(CH3CN)4PF6
1 Me2PhSiZnCl·2LiCl
(2.0 equiv.)
56 18
2 (Me2PhSi)2Zn·xLiCl
(1.2 equiv.)
36 0
3 (Me2PhSi)2Zn·xLiCl
(1.2 equiv.)
53 (6% ee)[c]
w/ L1
–
4[d] (Me2PhSi)2Zn·xLiCl
(1.2 equiv.)
55 (15% ee)[c]
w/ L1
–
[a] All reactions were performed on a 0.2 mmol scale for 2 h. [b]
Determined by 1H NMR spectroscopy by using CH2Br2as the internal
standard. [c] Determined by HPLC analysis on a chiral stationary phase. [d]
The 1,4-addition was conducted at 78°C for 16 h prior to the addition of
ClP(O)(OPh)2.
Chemistry—A European Journal
Communication
doi.org/10.1002/chem.202102993
16104Chem. Eur. J. 2021,27, 16103–16106 www.chemeurj.org © 2021 The Authors. Chemistry - A European Journal published by Wiley-VCH GmbH
Wiley VCH Donnerstag, 11.11.2021
2165 / 221299 [S. 16104/16106] 1
systematic screening of various chiral ligands was completely
unsuccessful (see the Supporting Information for the details).
However, the yield could be improved to 76% with no
enantioselectivity with (R,R,R)-L2 (see Scheme 3), and we
continued using this ligand for the reaction scope (a racemic
ligand such as rac-binap afforded significantly lower yields;
19% yield). For completion, the corresponding 1,4-addition of
Me2PhSiZnCl·2LiCl in the presence of (S,R,R)-L1 proceeded with
no enantioinduction.
The optimized reaction conditions are 5.0 mol% of Cu-
(CH3CN)4PF6and 6.0 mol% of L2 in THF with 1.2 equiv. of
(Me2PhSi)2Zn·xLiCl as the silicon source and ClP(O)(OPh)2as the
electrophilic trapping reagent (Scheme 3). The reaction scope
was done with the same set of cyclobutenones 1 a–p(cf.
Scheme 2). Yields were good throughout with β-aryl-substituted
cyclobutenones (1a–f!3a–f). Conversely, the β-alkyl-substi-
tuted derivatives were less reactive, and moderate yields were
obtained (1g–m!3g–m). Again, a silyl group as in 1n was
tolerated to give 3n in 52% yield. Both α,β-disubstituted
substrates 1o and 1 p did react in acceptable yields, affording
fully substituted enol phosphates 3o and 3p, respectively.
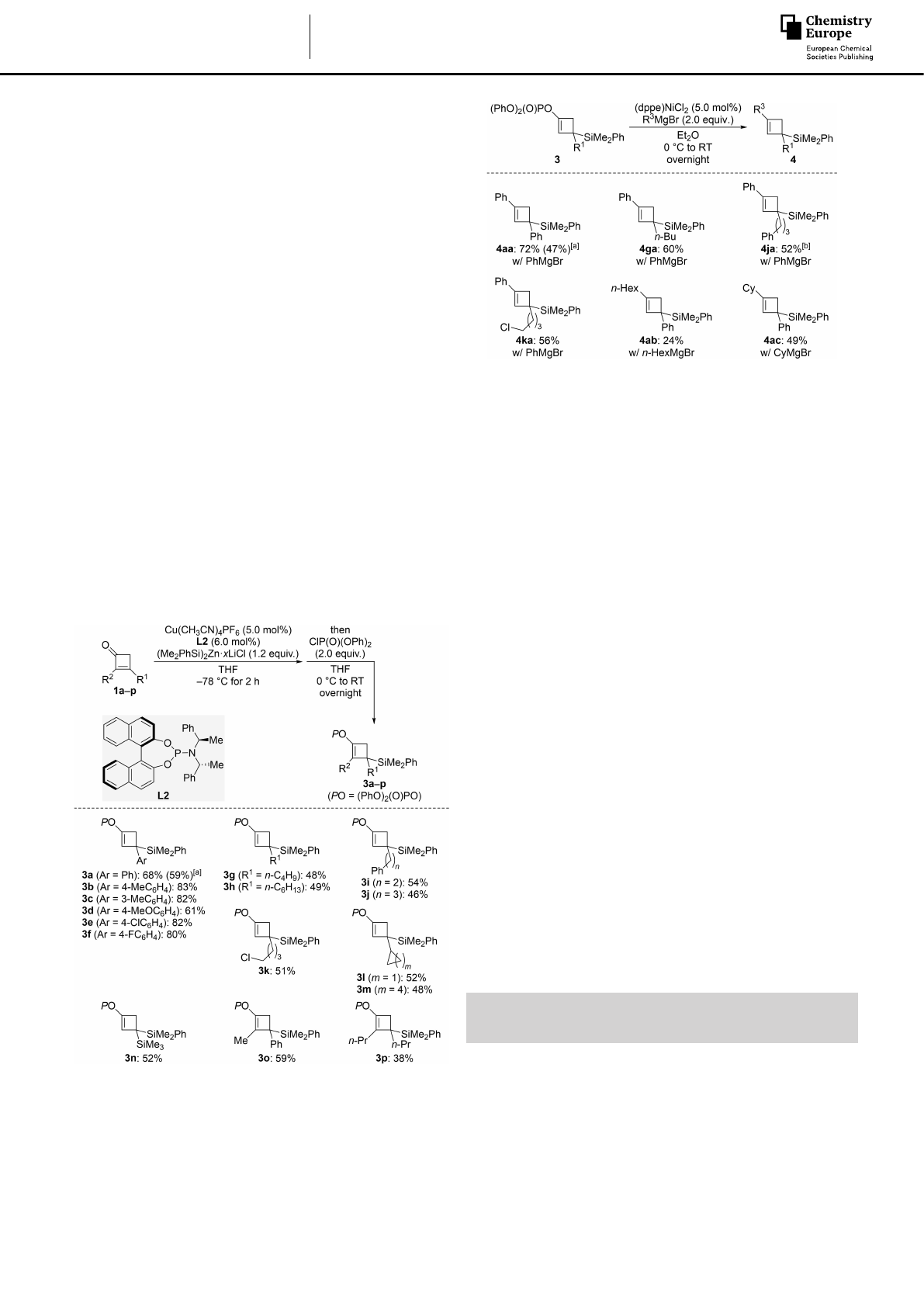
Enol phosphates can serve as electrophiles in cross-coupling
reactions,[15] and we tested several of the above cyclobutenyl
phosphates in Kumada coupling reactions (3!4, Scheme 4).
These representative reactions proceeded in moderate yields in
the presence of catalytic amounts of (dppe)NiCl2.[18] Arylation
with PhMgBr reliably gave the corresponding silylated cyclo-
butenes. In turn, alkylation with the primary alkyl Grignard
reagent n-HexMgBr was low yielding but an acceptable yield
was restored with secondary CyMgBr.
To summarize, we reported here a copper-catalyzed con-
jugate addition of zinc-based silicon reagents to highly
substituted cyclobutenones, providing a general and efficient
method to access various β-silylated cyclobutanones. Moreover,
the enolate intermediate can be trapped with a phosphorus
electrophile to arrive at silylated enol phosphates, and these
can be converted into the corresponding cyclobutenes by
Kumada cross-coupling.
Acknowledgements
M.C. thanks the China Scholarship Council for a predoctoral
fellowship (2018–2022). M.O. is indebted to the Einstein
Foundation Berlin for an endowed professorship. Open Access
funding enabled and organized by Projekt DEAL.
Conflict of Interest
The authors declare no conflict of interest.
Keywords: conjugate addition ·copper ·silicon ·synthetic
methods ·zinc
[1] a) E. Hartmann, D. J. Vyas, M. Oestreich, Chem. Commun. 2011,47,
7917–7932; for a recent Outlook, see: b) W. Xue, M. Oestreich, ACS Cent.
Sci. 2020,6, 1070–1081.
[2] F. Nahra, O. Riant in Science of Synthesis: Knowledge Updates 2013/2 (Ed.:
M. Oestreich), Thieme, Stuttgart, 2013, pp. 197–214.
[3] G. R. Jones, Y. Landais, Tetrahedron 1996,52, 7599–7662.
Scheme 3. Synthesis of silylated cyclobutenyl phosphates by sequential
conjugate addition of (Me2PhSi)2Zn·xLiCl (x�4) and enolate trapping. Unless
otherwise noted, all reactions were performed on a 0.2 mmol scale. Yields
are of analytically pure product obtained after flash chromatography on
silica gel. [a] Value in parentheses for the reaction on a 1.5 mmol scale.
Scheme 4. Nickel-catalyzed Kumada cross-coupling of silylated cyclobutenyl
phosphates and Grignard reagents. Unless otherweise noted, all reactions
were performed on a 0.10 mmol scale. Yields are of analytically pure product
obtained after flash chromatography on silica gel. [a] Value in parentheses
for the reaction on a 1.0 mmol scale. [b] Performed on a 0.065 mmol scale.
Chemistry—A European Journal
Communication
doi.org/10.1002/chem.202102993
16105Chem. Eur. J. 2021,27, 16103–16106 www.chemeurj.org © 2021 The Authors. Chemistry - A European Journal published by Wiley-VCH GmbH
Wiley VCH Donnerstag, 11.11.2021
2165 / 221299 [S. 16105/16106] 1

[4] a) D. J. Ager, I. Fleming, J. Chem. Soc. Chem. Commun. 1978, 177–178;
b) D. J. Ager, I. Fleming, S. K. Patel, J. Chem. Soc. Perkin Trans. 1 1981,
2520–2526; c) B. H. Lipshutz, J. A. Sclafani, T. Takanami, J. Am. Chem.
Soc. 1998,120, 4021–4022.
[5] a) C. Walter, G. Auer, M. Oestreich, Angew. Chem. Int. Ed. 2006,45, 5675–
5677; Angew. Chem. 2006,118, 5803–5805; b) C. Walter, R. Fröhlich, M.
Oestreich, Tetrahedron 2009,65, 5513–5520; c) K.-s. Lee, A. H. Hoveyda,
J. Am. Chem. Soc. 2010,132, 2898–2900.
[6] M. Murakami, Y. Miyamoto, Y. Ito, J. Am. Chem. Soc. 2001,123, 6441–
6442.
[7] M. Oestreich, B. Weiner, Synlett 2004, 2139–2142.
[8] G. Auer, B. Weiner, M. Oestreich, Synthesis 2006, 2113–2116.
[9] A. Weickgenannt, M. Oestreich, Chem. Eur. J. 2010,16, 402–412.
[10] H. Ito, T. Toyoda, M. Sawamura, J. Am. Chem. Soc. 2010,132, 5990–5992.
[11] C. K. Chu, Y. Liang, G. C. Fu, J. Am. Chem. Soc. 2016,138, 6404–6407.
[12] W. Xue, R. Shishido, M. Oestreich, Angew. Chem. Int. Ed. 2018,57,
12141–12145; Angew. Chem. 2018,130, 12318–12322.
[13] L. Zhang, M. Oestreich, Org. Lett. 2018,20, 8061–8063.
[14] C. Zhong, Y. Huang, H. Zhang, Q. Zhou, Y. Liu, P. Lu, Angew. Chem. Int.
Ed. 2020,59, 2750–2754; Angew. Chem. 2020,132, 2772–2776.
[15] Y. Nassar, F. Rodier, V. Ferey, J. Cossy, ACS Catal. 2021,11, 5736–5761.
[16] A solvent change from THF to Et2O was done to precipitate and remove
as much LiCl as possible. As we did not quantify the residual LiCl, we
decided to continue with Me2PhS2Zn·xLiCl (x�4). C. Fopp, E. Romain, K.
Isaac, F. Chemla, F. Ferreira, O. Jackowski, M. Oestreich, A. Perez-Luna,
Org. Lett. 2016,18, 2054–2057.
[17] L. Zhang, M. Oestreich, ACS Catal. 2021,11, 3516–3522.
[18] A. S. E. Karlström, K. Itami, J.-E. Bäckvall, J. Org. Chem. 1999,64, 1745–
1749.
Manuscript received: August 16, 2021
Accepted manuscript online: September 7, 2021
Version of record online: October 7, 2021
Chemistry—A European Journal
Communication
doi.org/10.1002/chem.202102993
16106Chem. Eur. J. 2021,27, 16103–16106 www.chemeurj.org © 2021 The Authors. Chemistry - A European Journal published by Wiley-VCH GmbH
Wiley VCH Donnerstag, 11.11.2021
2165 / 221299 [S. 16106/16106] 1
Loading more pages...