
Li-S Batteries
Origin and Acceleration of Insoluble Li2S2
Li2S Reduction Catalysis
in Ferromagnetic Atoms-based Lithium-Sulfur Battery Cathodes
Rui Yan+, Zhenyang Zhao+, Menghao Cheng, Zhao Yang, Chong Cheng,* Xikui Liu,*
Bo Yin, and Shuang Li*
Abstract: Accelerating insoluble Li2S2Li2S reduction
catalysis to mitigate the shuttle effect has emerged as an
innovative paradigm for high-efficient lithium-sulfur
battery cathodes, such as single-atom catalysts by
offering high-density active sites to realize in situ
reaction with solid Li2S2. However, the profound origin
of diverse single-atom species on solid-solid sulfur
reduction catalysis and modulation principles remains
ambiguous. Here we disclose the fundamental origin of
Li2S2Li2S reduction catalysis in ferromagnetic ele-
ments-based single-atom materials to be from their spin
density and magnetic moments. The experimental and
theoretical studies disclose that the FeN4-based cath-
odes exhibit the fastest deposition kinetics of Li2S
(226 mAhg1) and the lowest thermodynamic energy
barriers (0.56 eV). We believe that the accelerated
Li2S2Li2S reduction catalysis enabled via spin polar-
ization of ferromagnetic atoms provides practical oppor-
tunities towards long-life batteries.
Introduction
Lithium-sulfur (LiS) batteries have been regarded as the
most promising energy storage system due to the high
theoretical capacity (1675 mAhg1) and natural abundance
of sulfur element.[1] The high-energy and long-life LiS
batteries rely on the cathodes with efficient polysulfide
redox capability. In typical polysulfide redox chemistry, the
sulfur reduction reaction (SRR) undergoes a complex
conversion process from the sulfur molecule (S8) to soluble
Li2Sx(LiPSs, 4�x�8), ultimately generating insoluble Li2S2
and Li2S.[2] Fundamentally, the inherent sluggish SRR
kinetics result in low sulfur utilization and shuttle effect of
the soluble polysulfides.[3] This has led to two associated
trends in recent cathode design of LiS batteries: catalytic
sites with sufficient adsorption/bonding capability to poly-
sulfides and fast catalytic conversion of polysulfide inter-
mediates.
Theoretical calculations suggest that the rate-determin-
ing step in most of the SRR processes is the solid-solid
conversion from Li2S2to Li2S due to the sluggish solid
diffusion and poor interface contact between catalysts and
Li2S2.[4] Therefore, the sluggish electrodeposition of Li2S and
the associated accumulation of Li2Sxand dead sulfur have
long been considered as the root cause for the rapid capacity
fading of cathodes.[5] The key point to overcoming this
sluggish process relies on a strategy that weakens the SS
bond and promotes the Li2S2dissociation to accelerate the
insoluble Li2S2Li2S reduction catalysis.[6] However, the
influences and roles of diverse polysulfide redox catalysts on
Li2S2dissociation remain unclear, which is of great impor-
tance to be discovered for the future design of efficient and
long-cycling LiS battery cathodes.[7]
Due to the much less molecular movement ability in the
solid phase than that in solution, catalytic sites with high
activity and density are needed for the polysulfide redox
materials to realize an efficient in situ reaction with solid
Li2S2. Therefore, to accelerate the solid-solid conversion
kinetics, both geometric and electronic structures of the
cathode materials should be considered. Single-atom cata-
lysts (SACs), comprising monodispersed metal active sites
offer a theoretical 100% atom utilization, therefore, will
form an atomic-level contact/catalytic interface for promot-
ing solid-state Li2S2/Li2S conversion.[8] Recently, diverse
“SACs” cathodes have been reported in enhancing LiS
battery performances, for instance, the ferromagnetic ele-
ments (Fes=Fe, Co, and Ni)-based cathodes with metal-N4
structures have been demonstrated to possess enhanced
polysulfide catalytic conversion ability.[9] Regrettably, the
insoluble Li2S2Li2S reduction mechanisms via taking the
FEs-based SACs is unclear, and the corresponding correla-
tion between the catalytic activities and electronic structures
of FEsN4remain undiscovered.
In this work, we provide a comparative study on the
fundamental origin of insoluble Li2S2Li2S reduction catal-
[*] R. Yan,+Z. Zhao,+M. Cheng, Z. Yang, Prof. C. Cheng, Prof. X. Liu,
Prof. B. Yin, Prof. S. Li
College of Polymer Science and Engineering, State Key Laboratory
of Polymer Materials Engineering, Sichuan University
Chengdu 610065 (China)
E-mail: [email protected]
Prof. S. Li
Department of Chemistry, Technische Universität Berlin
Berlin 10623 (Germany>)
E-mail: [email protected]
[+] These authors contributed equally to this work.
© 2022 The Authors. Angewandte Chemie International Edition
published by Wiley-VCH GmbH. This is an open access article under
the terms of the Creative Commons Attribution License, which
permits use, distribution and reproduction in any medium, provided
the original work is properly cited.
Angewandte
Chemie
Research Articles www.angewandte.org
How to cite: Angew. Chem. Int. Ed. 2023, 62, e202215414
International Edition: doi.org/10.1002/anie.202215414
German Edition: doi.org/10.1002/ange.202215414
Angew. Chem. Int. Ed. 2023,62, e202215414 (1 of 10) © 2022 The Authors. Angewandte Chemie International Edition published by Wiley-VCH GmbH

ysis in FEs-based SAC cathodes with different metal-N4
sites. Through a series of theoretical studies, we disclose that
the spin polarization (FeN4>CoN4>NiN4) can provide
spin electrons to reduce antibonding orbitals occupation in
Li2S2FEsN4and enhance the FEsS interaction, thereby
weakening the strength of the SS bond in Li2S2, and
eventually accelerating the Li2S2Li2S reduction catalysis at
cathode interface. Meanwhile, we have synthesized a series
of FEs-based single-atom sites loaded on hierarchical porous
carbon (HP-SAFEs) as cathode materials to verify the
proposed mechanism. Thereafter, systematically spectro-
scopic, structural, and electrochemical studies have demon-
strated that the FeN4-based cathodes exhibit the fastest
Li2S2Li2S reduction kinetics and the highest capacity
retention of 578 mAhg1after 200 cycles under 1 C (1 C=
1675 mAg1), which is far exceeding those of HP-SACo
(512 mAhg1) and HP-SANi (454 mAhg1) based batteries.
Our findings suggest that the spontaneous spin polarization
of ferromagnetic atoms can accelerate insoluble Li2S2Li2S
reduction catalysis, thus offering a new strategy to design
high-energy and long-life polysulfide reduction catalysts for
practical LiS batteries.
Results and Discussion
To disclose the origin of insoluble Li2S2Li2S reduction
catalysis for the FEs-based SAC cathodes with different
metal-N4sites, the adsorption and dissociation free energies
of Li2S2are calculated by taking the density function theory
(DFT) method. It has been reported that the dorbitals of
transition metal centers play a crucial role in the interaction
with reaction intermediates.[10] The change of dorbitals can
modify the related electronic structures, which in turn affect
the reaction energy barrier. For the polysulfide reduction
catalysts, the effective electronic states of intermediates
(Li2S2and Li2S) are usually dominated by the porbitals of
sulfur.[11] The hybridization between dorbitals of metal
centers and porbitals of sulfur will largely affect the
catalytic activities. Therefore, the d-p orbital hybridization
between different FEs-based SACs and Li2S2are explored
to predict their reaction free energies. The partial projected
density of state (PDOS) of the FEsN4(Figure S1, Support-
ing Information) shows that the electronic states of d
orbitals (FeN4) present an asymmetry that originated from
the uneven distribution of electrons in spin up and spin
down. In comparison, this asymmetry of dorbitals in CoN4
is weaker and eventually disappears completely in the NiN4
center. The asymmetry of dorbitals for FEsN4will cause
spontaneous spin polarization that can provide spin elec-
trons, which is beneficial for bonding with polysulfide
molecules.[9b,12] In order to quantify the spin polarization of
FEs-based SAC, their spin density and magnetic moments
are calculated. Figure 1a and Figure S2 (Supporting Infor-
mation) shows that FeN4possessed the largest spin density
and magnetic moment of 1.91 μB, suggesting its superior spin
polarization degree compared to the CoN4and NiN4.
To confirm that the ability of chemisorbing polysulfides
is related to the spin polarization degree, we further analyze
the molecular orbitals of Li2S2, FEsN4, and Li2S2FEsN4
(Figure S3, S4, Supporting Information). Results show that
among FEsN4, the FeN4possessing most spin electrons,
thus leading to less antibonding orbitals occupation in
Li2S2FeN4and resulting in robust FeS interaction.
Furthermore, the optimized adsorption configuration of
Li2Sx(1�x�8) on FEsN4is considered (Figure S5, S6,
Supporting Information). In the case of FeN4and CoN4,
Li bond with the N/C atom and S bond with Fe or Co atom,
respectively. While for the NiN4, there is no apparent
interaction between S and Ni atoms. Different from Ni-
based compounds, it is the LiN instead of SNi interaction
promoting NiN4absorbing Li2Sx. The binding strength of
Li2Sxwith FEsN4sites (Figure 1b) shows that the FeN4
has the highest binding energies with S8, Li2S6, and Li2S2of
0.96, 1.28, and 1.81 eV, respectively, in agreement with
the results of the magnetic moment and spin polarization
degree.
Considering that the conversion from Li2S2to Li2S
involves the dissociation of the SS bond in Li2S2, the PDOS
of Li2S2FEsN4has been analyzed to reveal the relation-
ship between the adsorption/reduction of Li2S2on FEsN4
surface and the electronic structure of the SACs. When the
electronic states of FEs interact with the S atom, the
hybridized energy levels will split into the anti-bonding
states (normally go across the Fermi level (Ef)) and the
bonding states (below the Ef) (Figure S7, Supporting
Information). The strength of the FEsS interaction
depends on the position of the antibonding state, and the
higher the position of the anti-bonding state, the stronger
the interaction. As shown in Figure 1c, the d-band centers
(ɛd) of FEsN4exhibit ɛd(Fe) (0.79 eV)>ɛd(Co) (1.74 eV)>
ɛd(Ni) (2.32 eV), meaning the ɛd(Fe) in Li2S2FeN4is closer
to the Fermi level. This leads to a stronger interaction
between Li2S2and FeN4than the Li2S2CoN4and
Li2S2NiN4, which corresponds to the order of adsorption
energy, thus consequently weakening the SS bonds in
Li2S2.[13] Meantime, the Li2S2FeN4shows more electron
occupation on its dorbitals near the Fermi level makes it
more prone to accept or lose electrons, which suggests an
excellent electron transfer ability of Fe sites and offers
benefit to the following Li2S2reduction reaction.[14]
Furthermore, an obvious charge redistribution between
the Li2S2and FEsN4can be observed in the diagrams of
charge density differences. The number of charge transfers
between the FEs and S bond has been calculated by Bader
charge analysis (Figure 1d–f); there are 0.20, 0.18, and 0 jej
charges that transfer from S atom to FEs atoms, respec-
tively, suggesting the strongest electron exchange between S
and Fe atoms. To further learn the actual states of SS
interaction, the PDOS of S in Li2S2FEsN4is analyzed. As
shown in Figure 1g, the (s p) orbitals of S2 (the S that
directly connects to Fe) match well with S1 in Li2S2NiN4,
indicating a strong SS bond. This degree of matching is
weakened in Li2S2CoN4and Li2S2FeN4, where the latter
is particularly pronounced, implying a weakened SS bond
due to the transfer of internal charge from the S2 to Fe site.
We then calculate the bond lengths of SS and FEsS in
Li2S2FEsN4to reveal their internal interactions (Fig-
Angewandte
Chemie
Research Articles
Angew. Chem. Int. Ed. 2023,62, e202215414 (2 of 10) © 2022 The Authors. Angewandte Chemie International Edition published by Wiley-VCH GmbH
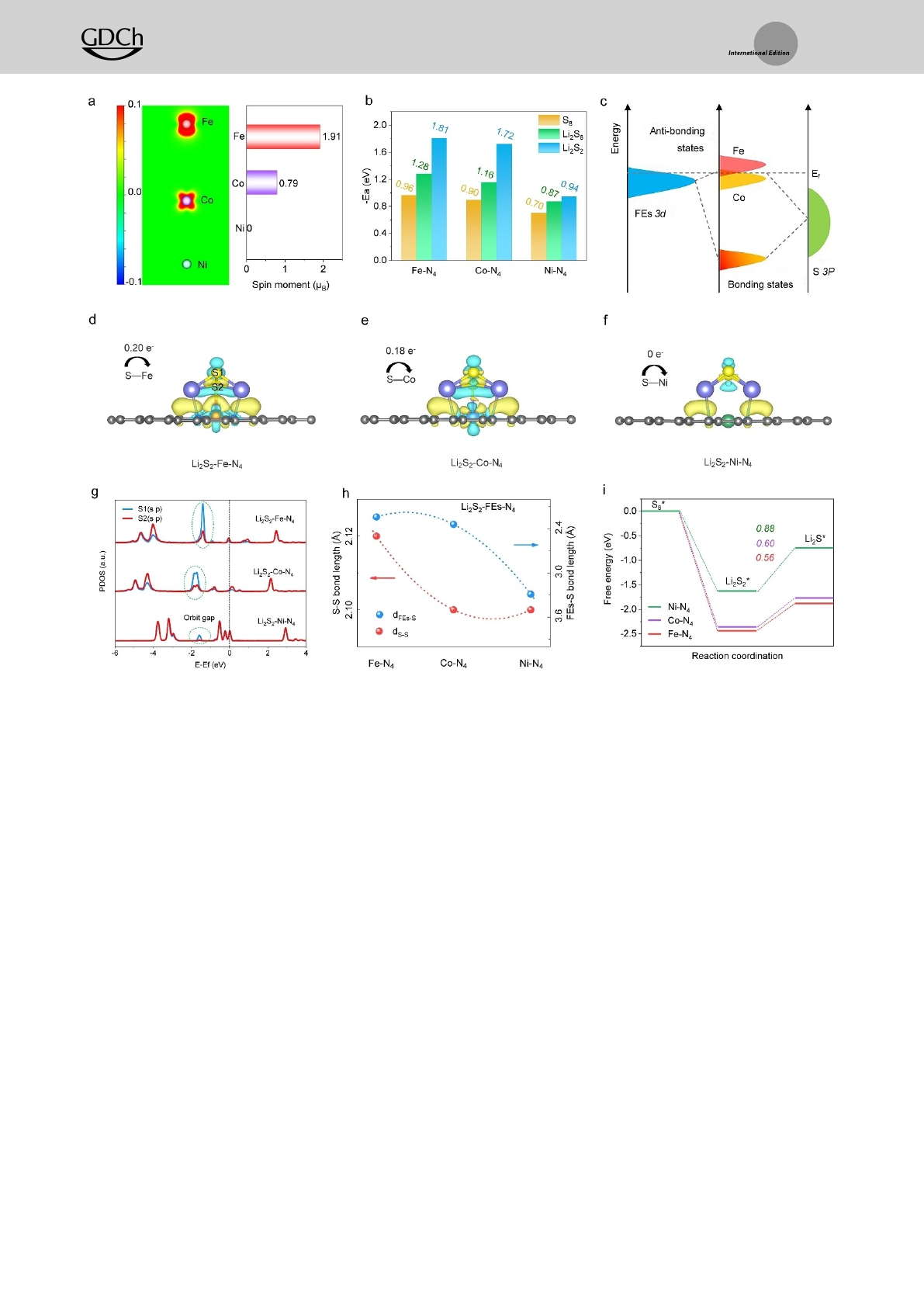
ure 1h). Notably, the SS bond lengths show a trend of
FeN4(2.120 Å)>CoN4(2.103 Å)>NiN4(2.101 Å), while
the FEsS bond lengths present an order of dFeS(2.24 Å)<
dCoS(2.31 Å)<dNiS(3.29 Å), thus indicating a strong
interaction and electron transfer within FeS, therefore
effectively weakening the SS bond in Li2S2. The free energy
diagrams for the Li2S2Li2S reduction catalysis in FEs-based
SAC show that the FeN4site (0.56 eV) displays the lowest
thermodynamic energy barrier compared to the CoN4
(0.60 eV) and NiN4(0.88 eV), respectively (Figure 1i). All
the calculation results reveal that the FeN4catalyst
presents the easiest Li2S2Li2S reduction conversion activity,
indicating that the spin polarization is responsible for its
enhanced FEsS interaction and accelerated solid-solid
catalytic conversion, which will also be further confirmed by
our following experimental results.
To verify the above-proposed mechanism, we have
synthesized a series of HP-SAFEs (Figure 2a) as Li2S2Li2S
reduction catalysts to assemble LiS batteries by utilizing
silica embedded nanocubic metal-organic precursor with in
situ FEs doping (Figure S8, Supporting Information). First,
the precursors are pyrolyzed at 900°C in the Ar atmosphere;
after removing silica, secondary thermal treatment is con-
ducted to obtain the HP-SAFEs. All the synthesized HP-
SAFEs exhibit cubic morphologies with rough surfaces
(Figure S9, Supporting Information); meantime, obvious
mesoporous are found under scanning electron microscopy
(SEM). The control sample of hierarchical porous N-doped
carbon without metal atoms doping (HP-NC) is also
prepared. The similar specific surface areas and pore
structures of the HP-SAFe (1079 m2g1), HP-SACo
(1002 m2g1), HP-SANi (1056 m2g1), and HP-NC
(1130 m2g1) are validated by the N2adsorption/desorption
analysis (Figure 2b and c, Figure S10, Supporting Informa-
tion).
To further explore the electronic structure and coordina-
tion environment of HP-SAFEs, we first analyze the high-
resolution X-ray photoelectron spectroscopy (XPS) of N 1s
Figure 1. Theoretical understanding for the Li2S2
Li2S reduction catalysis in FEs-based SAC. a) The calculated spin density and magnetic moment
of FEsN4. b) The binding energy of S8, Li2S6, and Li2S2with FEsN4. c) Schematic illustration of the local electronic structure of Fe/Co 3d-orbitals
in Li2S2
FEsN4, Efis the Fermi level. The charge density differences of d) Li2S2
FeN4, e) Li2S2
CoN4, and f) Li2S2
NiN4. g) The calculated
PDOS of S in Li2S2
FeN4, Li2S2
CoN4, and Li2S2
NiN4. h) The bond lengths of FEsS and SS in Li2S2
FEsN4, respectively. i) The free energy
diagrams for the reduction process of Li2S2to Li2S on the FEsN4.
Angewandte
Chemie
Research Articles
Angew. Chem. Int. Ed. 2023,62, e202215414 (3 of 10) © 2022 The Authors. Angewandte Chemie International Edition published by Wiley-VCH GmbH
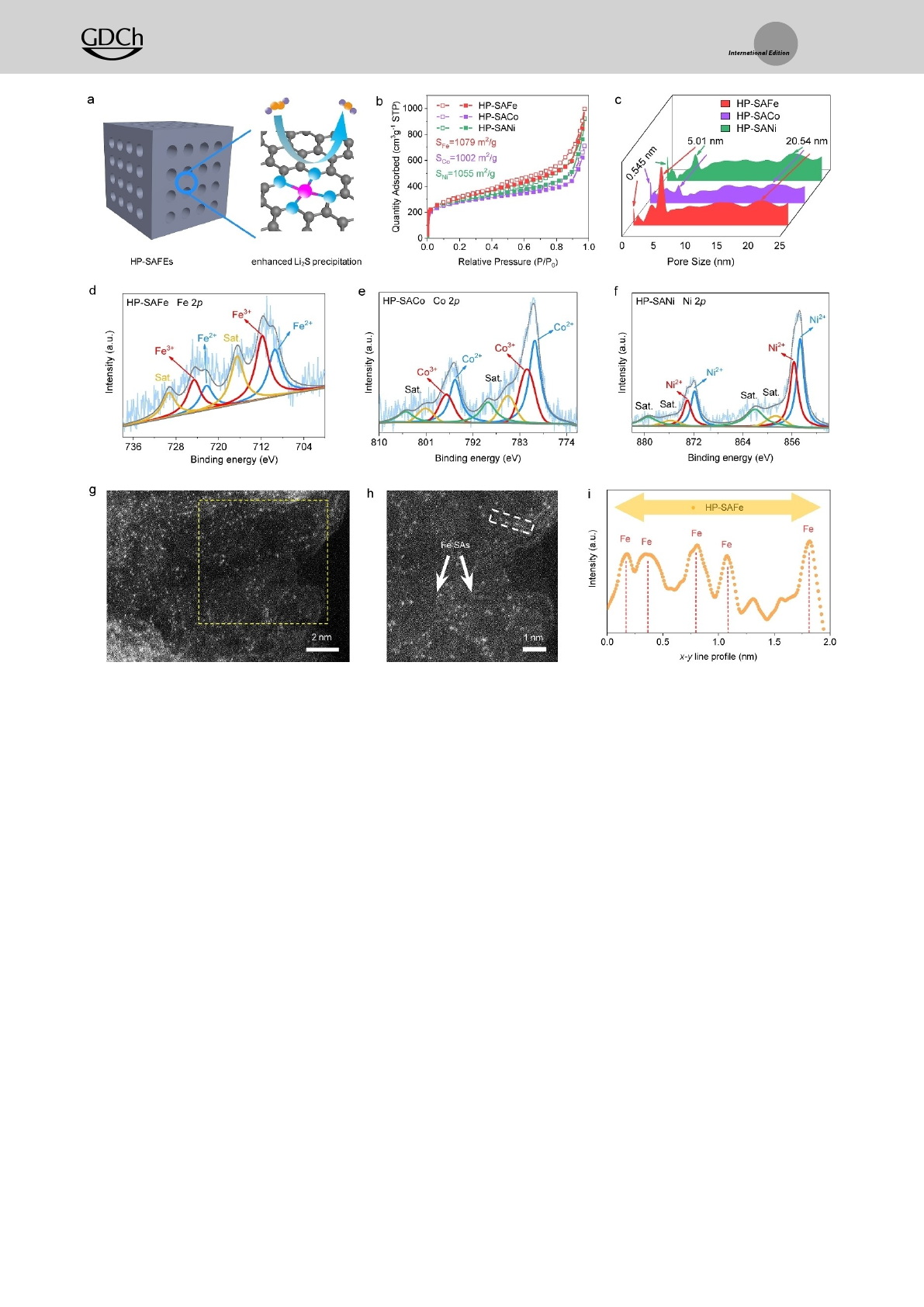
spectra. The result shows that pyridinic N in HP-NC has a
binding energy of 398.35 eV, while it shifts to 398.55 eV for
HP-SAFEs (Figure S11, Supporting Information), which
suggests that the metal ions are bonded with pyridinic N to
form the atomic metal-Nxsites.[15] Moreover, the XPS and
inductively coupled plasma (ICP) confirms that HP-SAFEs
display similar metal contents of Fe (0.26 At.%, 1.09 wt%),
Co (0.42 At.%, 1.15 wt%), and Ni (0.34 At.%, 0.85 wt%),
respectively (Figure 2d–f, Table S2, Supporting Informa-
tion). The Fe region scan shows two main peaks in the Fe
2p3/2 at the binding energy of 709.20 and 711.70 eV, which
correspond to the Fe2+and Fe3+oxidation states, respec-
tively. Ni 2p and Co 2p spectra also present a similar
oxidation state of Co2+/Co3+(780.13 and 781.60 eV) and
Ni2+(854.66 and 855.67 eV).[16] No zero-valent metal peaks
can be found for all the XPS spectra of HP-SAFEs, thus
indicating no metallic particles or clusters in these HP-
SAFEs.
The atomic-scale structure of the representative HP-
SAFe is future observed under a spherical aberration-
corrected scanning transmission electron microscope (AC-
STEM). Figure 2g–i and Figure S14 (Supporting Informa-
tion) show abundant bright dots, indicating the atomic
distribution of Fe atoms in the porous carbon substrate; no
Fe clusters or particles can be observed. The X-ray
absorption near-edge structure (XANES) and extended X-
ray absorption fine structure (EXAFS) spectroscopy are
performed to further reveal the coordination environment
and valence state of Fe atoms. The XANES curves at the Fe
K-edge show that the position of HP-SAFe is located
between those of Fe3O4and Fe2O3, corroborating the
valence state is between Fe2+and Fe3+, meantime HP-SACo
corroborates the valence state between Co2+and Co3+, and
HP-SANi between Ni0and Ni2+(Figure 3a–c). Fourier-trans-
forms (FTs) and wavelet-transforms (WTs) images from the
EXAFS spectra of HP-SAFEs depict that the FEs is found
to be bonded as FEsN/C/O.[17] No obvious metal peak in
Figure 2. Structure analysis of HP-SAFEs. a) The schematic porous structure and b) N2adsorption-desorption measurements and c) corresponding
pore size distributions for HP-SAFEs. d) XPS spectra of Fe 2p for HP-SAFe, e) Co 2p for HP-SACo, and f) Ni 2p for HP-SANi. g) AC-STEM image
and h) the magnified image of HP-SAFe. i) x-y line scan profile, measured from (h). j) XANES spectra at the Fe K-edge and k) their magnified
image. l) Fourier transforms the EXAFS spectra for Fe K-edge of Fe foil, Fe2O3, FePc, and HP-SAFe. m) The corresponding EXAFS fitting curves of
HP-SAFe at R space. Wavelet transformation of Fe K-edge EXAFS of n) Fe2O3, o) HP-SAFe, and p) FePc, respectively.
Angewandte
Chemie
Research Articles
Angew. Chem. Int. Ed. 2023,62, e202215414 (4 of 10) © 2022 The Authors. Angewandte Chemie International Edition published by Wiley-VCH GmbH
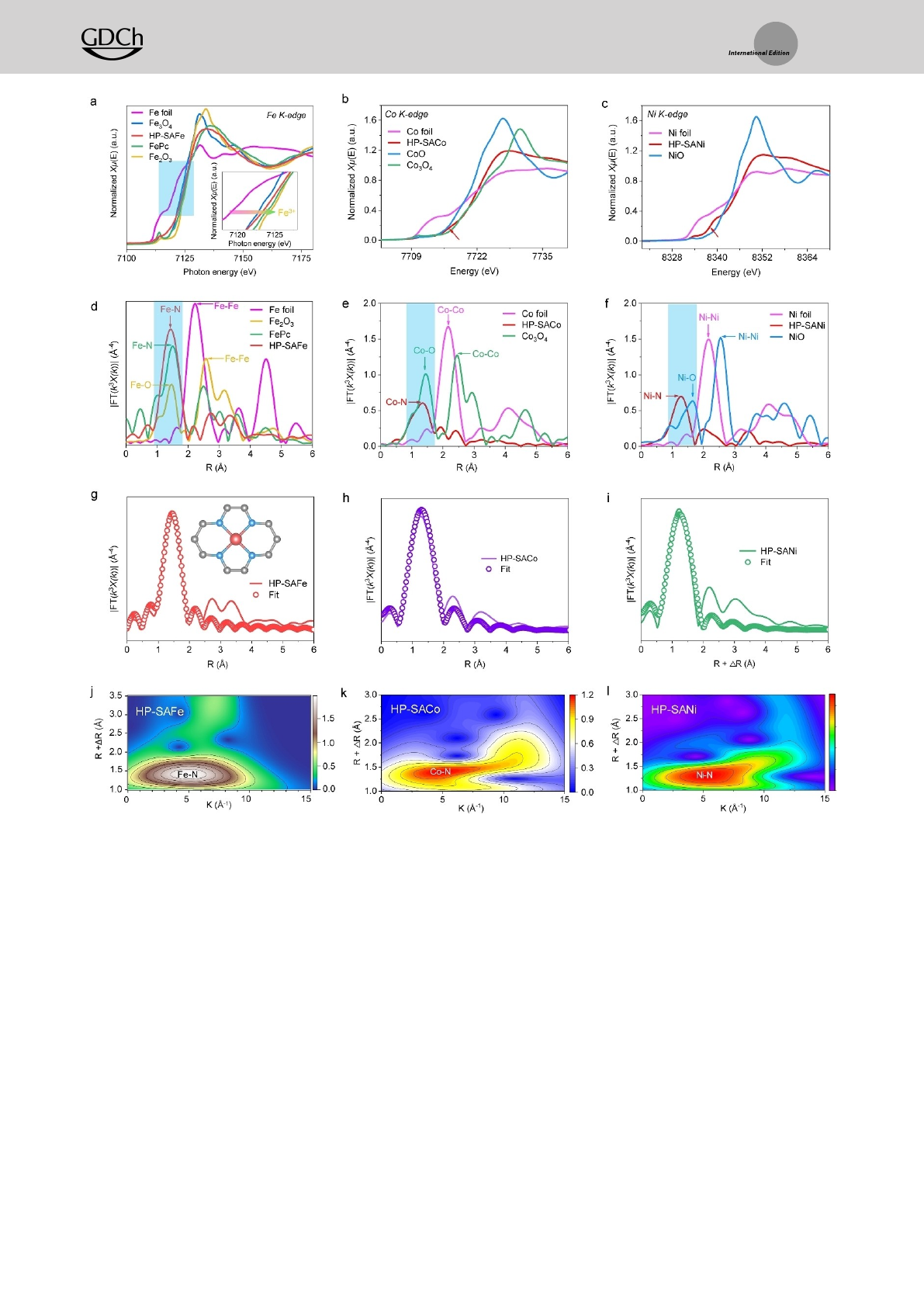
the FTs spectrum of HP-SAFEs is observed, revealing their
atomic dispersion (Figure 3d–l). The least-squares EXAFS
fitting parameters at the Fe K-edge of HP-SAFe show the
FeN bond length of 1.99 Å and coordination number of
4.1, which are very similar to that determined for FePc
(2.01 Å, n=4.0) (Figure 3g, Figure S15 and Table S3, Sup-
porting Information). Meanwhile, HP-SACo and HP-SANi
show the CoN and NiN bond length of 1.75 Å and 1.71 Å
with coordination number of 4.0 and 4.1, respectively (Fig-
ure 3h and i). Based on the above analysis, we suggest that
the isolated FEs atoms in HP-SAFEs are tetra-coordination
by N atoms and form a typical FEsN4structure in the HP-
NC matrix.
After validation that the HP-SAFEs display similar
morphologies, surface areas, pore structures, and metal
contents, it is reliable to use these synthesized HP-SAFEs to
explore and compare the Li2S2Li2S reduction activities of
SAFEs experimentally. After heating the mixture of HP-
SAFEs and sulfur at 155°C, the resulted S@HP-SAFEs
containing 80 wt% sulfur (Figure S16, Supporting Informa-
tion) are used as cathodes in LiS batteries. The cubic
morphology of S@HP-SAFEs can be well-maintained after
sulfur is immitted (Figure S17, S18, Supporting Informa-
Figure 3. Configuration analysis of HP-SAFEs. a) XANES spectra at the Fe K-edge, b) Co K-edge, c) Ni K-edge. d) Fourier transforms the EXAFS
spectra for Fe K-edge of HP-SAFe, e) Co K-edge of HP-SACo, f) Ni K-edge of HP-SANi. g) The corresponding EXAFS fitting curves of HP-SAFe,
h) HP-SACo, i) HP-SANi at R space. Wavelet transformation of j) Fe K-edge EXAFS of HP-SAFe, k) Co K-edge EXAFS of HP-SACo, and l) Ni K-edge
EXAFS of HP-SANi, respectively.
Angewandte
Chemie
Research Articles
Angew. Chem. Int. Ed. 2023,62, e202215414 (5 of 10) © 2022 The Authors. Angewandte Chemie International Edition published by Wiley-VCH GmbH
Loading more pages...