
SPECIAL ISSUE - RESEARCH ARTICLE
Resonance Raman spectroscopic analysis of the iron–sulfur
cluster redox chain of the Ralstonia eutropha membrane-
bound [NiFe]-hydrogenase
Elisabeth Siebert
1
| Andrea Schmidt
2
| Stefan Frielingsdorf
1
|
Jacqueline Kalms
2,3
| Uwe Kuhlmann
1
| Oliver Lenz
1
| Patrick Scheerer
2
|
Ingo Zebger
1
| Peter Hildebrandt
1
1
Institute of Chemistry, Technical University of Berlin, Berlin, Germany
2
Institute of Medical Physics and Biophysics (CC2), Charité—University Medicine Berlin, Free University of Berlin, Humboldt University of Berlin,
Berlin, Germany
3
Leicester Institute of Structural and Chemical Biology, University of Leicester, Leicester, UK
Correspondence
Patrick Scheerer, Charité—
Universitätsmedizin Berlin, Freie
Universität Berlin, Humboldt-Universität
zu Berlin, Institute of Medical Physics and
Biophysics (CC2), Charitéplatz 1, D-10117
Berlin, Germany.
Email: [email protected]e
Ingo Zebger and Peter Hildebrandt,
Institut für Chemie, Technische
Universität Berlin, Sekr. PC14, Straße des
17. Juni 135, D-10623 Berlin, Germany.
Email: [email protected];
Funding information
European Union; European Union, Grant/
Award Number: 810856; Einstein
Foundation Berlin; Deutsche
Forschungsgemeinschaft; European
Synchrotron Radiation Facility; Helmholtz
Zentrum Berlin für Materialien und
Energie
Abstract
Iron–sulfur (Fe–S) centers are versatile building blocks in biological electron
transfer chains because their redox potentials may cover a wide potential range
depending on the type of the cluster and the specific protein environment. Reso-
nance Raman (RR) spectroscopy is widely used to analyze structural properties
of such cofactors, but it remains still a challenge to disentangle the overlapping
signals of metalloproteins carrying several Fe–S centers. In this work, we com-
bined RR spectroscopy with protein engineering and X-ray crystallography to
address this issue on the basis of the oxygen-tolerant membrane-bound hydroge-
nase from Ralstonia eutropha that catalyzes the reversible conversion of hydro-
gen into protons and electrons. Besides the NiFe-active site, this enzyme harbors
three different Fe–S clusters constituting an electron relay with a distal [4Fe–
4S], a medial [3Fe–4S], and an unusual proximal [4Fe–3S] cluster that may carry
a hydroxyl ligand in the superoxidized state. RR spectra were measured from
protein crystals by varying the crystal orientation with respect to the electric field
vector of the incident laser to achieve a preferential RR enhancement for individ-
ual Fe–S clusters. In addition to spectral discrimination by selective reduction of
the proximal cluster, protein engineering allowed for transforming the proximal
and medial cluster into standard cubane-type [4Fe–4S] centers in the C19G/
C120G and P242C variants, respectively. The latter variant was structurally char-
acterized for the first time in this work. Altogether, the entirety of the RR data
Elisabeth Siebert and Andrea Schmidt contributed equally to this work.
Received: 10 April 2021 Revised: 8 May 2021 Accepted: 9 May 2021
DOI: 10.1002/jrs.6163
This is an open access article under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in any medium, provided
the original work is properly cited.
© 2021 The Authors. Journal of Raman Spectroscopy published by John Wiley & Sons Ltd.
J Raman Spectrosc. 2021;52:2621–2632. wileyonlinelibrary.com/journal/jrs 2621

provided the basis for identifying the vibrational modes characteristic of the vari-
ous cluster states in this “model”enzyme as a prerequisite for future studies of
complex (FeS)-based electron transfer chains.
KEYWORDS
electron transfer, hydrogenase, iron–sulfur cluster, protein crystals, Raman spectroscopy
1|INTRODUCTION
Electron transfer chains in enzymes typically utilize an
array of cofactors that are spatially arranged in the pro-
tein structure to ensure an efficient electron transport
over long distances. In many cases, the electron relay is
based on iron–sulfur (Fe–S) centers, including two, three
or four Fe ions coordinated by cysteine residues and inor-
ganic sulfur atoms.
[1–5]
The redox potentials of these
cofactors are controlled by the type of the Fe–S cluster
and its specific interaction with the protein environment
and can span a wide potential range from 700 to
+450 mV (vs. NHE).
[6]
Thus, Fe–S clusters are versatile
cofactors adapted for fast electron transfer between pro-
tein sites of quite different potential levels.
To elucidate electron transfer processes along Fe–S
chains, the choice of methods is rather restricted. Unlike
other types of redox cofactors, UV–vis absorption spec-
troscopy, which is typically used for redox titrations, is
blind towards most reduced Fe–S clusters and can hardly
distinguish between the different cluster types.
[7]
Elec-
tron paramagnetic resonance (EPR) spectroscopy is
restricted to non-zero spin states and thus yields only an
incomplete picture about Fe–S clusters.
[8]
Mößbauer
spectroscopy is able to probe different spin and redox
states of Fe–S clusters and allows distinguishing different
structures.
[9]
However, application of this technique
requires
57
Fe labeling. Also resonance Raman
(RR) spectroscopy is associated with a drawback because
resonance enhancement of the vibrational modes
depends on a distinct electronic transition, limiting the
detection of Fe–S clusters in the oxidized state, similar to
UV–vis spectroscopy.
[10]
Unlike UV–vis spectroscopy,
however, RR spectroscopy provides a better spectral reso-
lution, which enables an unambiguous identification of
the cluster type. In fact, the Fe–S cluster manifold has
been the subject of a large number of RR spectroscopic
studies revealing characteristic vibrational signatures of a
large number of Fe–S centers, including [2Fe–2S], [3Fe–
4S], and [4Fe–4S] clusters.
[10,11]
Nevertheless, it remains
a formidable challenge to disentangle the RR spectra of
entire electron transfer chains in terms of their individual
components, even if each of them belongs to a different
Fe–S cluster type.
Here, we analyzed by RR spectroscopy the electron
transfer chain of the intensely studied oxygen-tolerant
membrane-bound [NiFe]-hydrogenase (MBH) from
Ralstonia eutropha (Figure 1). Our “model”enzyme cata-
lyzes the oxidation of molecular hydrogen into protons
and electrons at a Ni–Fe center, in which the two metal
ions are linked via two bridging cysteines. The Ni ion is
further coordinated by two cysteines whereas the Fe
carries additionally two cyanide and CO ligands. Efficient
oxidation of H
2
requires the rapid removal of electrons
from the active site, which is mediated by three different
Fe–S clusters, named proximal, medial, and distal cluster
according to their location relative to the catalytic center.
The medial and distal clusters are prototypical [3Fe–4S]
and [4Fe–4S] clusters, respectively. The proximal Fe–S
cluster, by contrast, exhibits a quite unusual structure.
[12–14]
Here, four iron ions are coordinated by three sulfides
and six cysteine-derived thiolate sulfurs, which enable
the [4Fe–3S] cluster to undergo redox-dependent struc-
tural and conformational changes. In contrast to conven-
tional Fe–S clusters, which usually switch between two
different redox states, the [4Fe–3S] cluster can adopt even
three redox states under physiological conditions. No
structural changes occur when the cluster switches
between the reduced and oxidized state. Transition from
the oxidized to the superoxidized state, however, is
accompanied with the exchange of the coordination of
one iron (named Fe4) from a sulfide ligand to the back-
bone nitrogen of a cysteine residue (Cys20). A second
cluster-bound Fe (named Fe1) becomes coordinated by a
hydroxyl ligand in the superoxidized form of the [4Fe–
3S] cluster.
[12]
This unique structural change has been
attributed to the cluster's capability to switch between
three redox states at physiological potentials and the
corresponding O
2
tolerance of the MBH, that is, its
unusual capacity to sustain catalysis under aerobic
conditions.
In this work, we employed a combined approach of
protein engineering, X-ray crystallography, and RR spec-
troscopy to determine the characteristic vibrational signa-
tures of the three Fe–S clusters of MBH. The RR
spectroscopic experiments were carried out on MBH crys-
tals at 77 K. First, RR spectra of native MBH crystals were
measured as a function of the crystal orientation with
2622 SIEBERT ET AL.
respect to the electric field vector of the incident laser
radiation. This strategy exploits the different orientations
of the transition dipole moments of the three Fe–S clus-
ters relative to the crystal axes. In this way, a preferential
enhancement of RR-active modes for the individual Fe–S
clusters can be achieved upon varying the angle of the
electric field vector of the incident laser light with respect
to the principal crystal long [c] axis of the needle-shaped
crystals. Second, additional spectroscopic information
was obtained by selective reduction of the proximal [4Fe–
3S] cluster. Third, further spectral discrimination was
achieved by a comparative analysis of native MBH with
MBH variants in which cluster-coordinating or adjacent
residues were exchanged. This included the previously
investigated C19G/C120G variant
[15]
and the P242C vari-
ant that was characterized for the first time in this work.
These exchanges revealed a four-cysteine coordination as
prototypical [4Fe–4S] cluster of both the proximal and
the medial Fe–S clusters.
2|MATERIALS AND METHODS
2.1 |Protein expression and
crystallization
Purification and crystallization of the heterodimeric
MBH from R. eutropha H16 was described previously.
[16]
The P242C variant of MBH was crystallized under aero-
bic and anaerobic conditions and the structures were
analyzed as described in detail in the supporting informa-
tion (section 1, Table S2). The crystals of all MBH vari-
ants studied in this work belong to the orthorhombic
space group P2
1
2
1
2
1
.
[14]
2.2 |RR spectroscopy
RR spectra were measured using a confocal Raman spec-
trometer (LabRam HR-800, Jobin Yvon) coupled to a
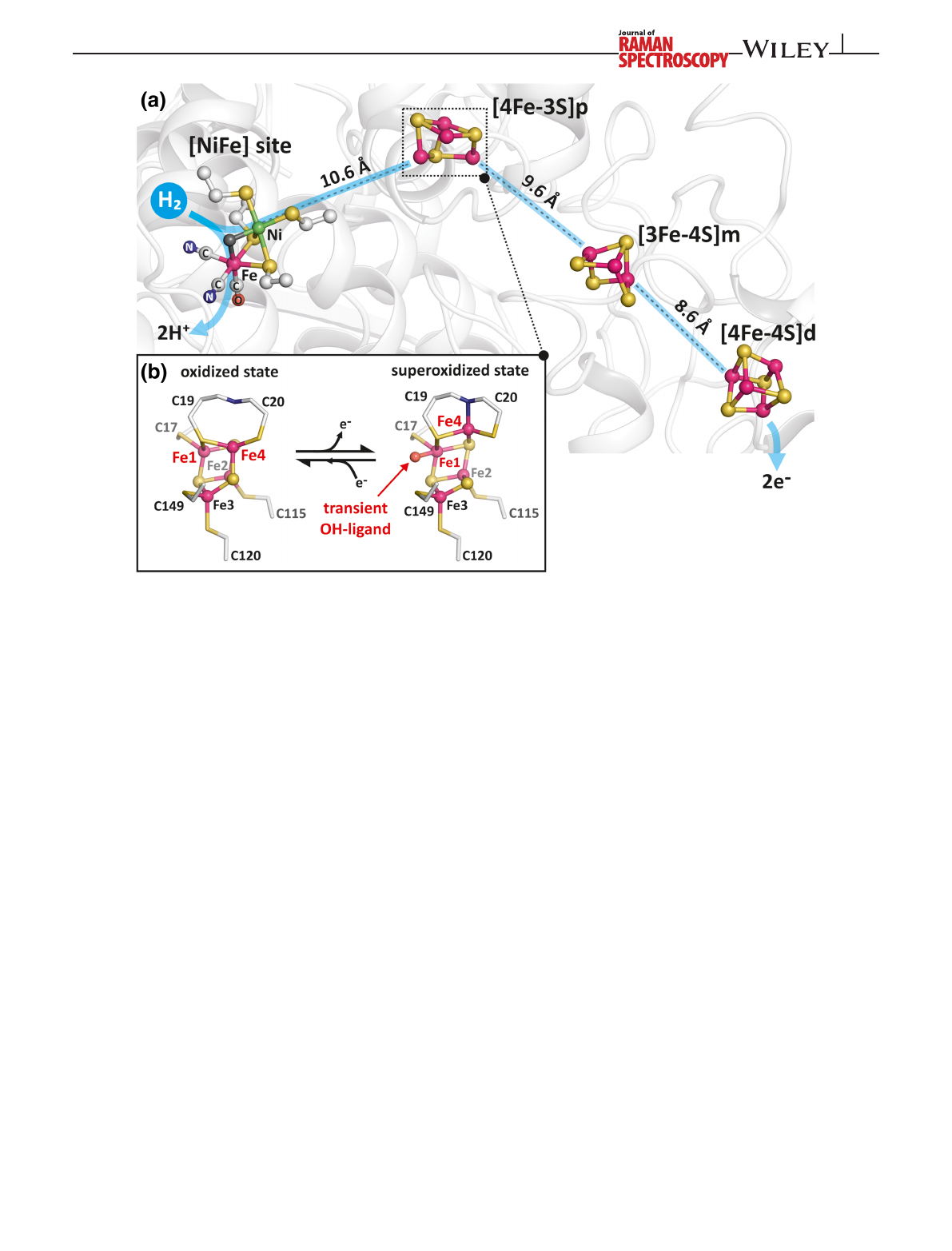
FIGURE 1 Schematic representation of the metal cofactors in the as-isolated native membrane-bound [NiFe]-hydrogenase (MBH):
(a) [NiFe] active site, proximal [4Fe–3S]p, medial [3Fe–4S]m, and distal [4Fe–4S]d clusters are represented as ball and sticks with their
distances from each other as found in the crystal structure.
[12]
(b) Close-up of the proximal [4Fe–3S]p in two redox states as oxidized and
superoxidized form. The superoxidized [4Fe–3S]p contains a transient Fe1-bound OH group
[12,13]
SIEBERT ET AL.2623
liquid nitrogen-cooled charge-coupled device (CCD). The
spectral resolution was limited by the wavenumber incre-
ments per pixel of the CCD camera, corresponding to
approximately 1.0 cm
1
for the excitation wavelength of
457 nm provided by an Ar ion laser (coherent). The laser
beam was focused on the sample surface by a Nikon 20
objective with a working distance of 20.5 mm and a
numeric aperture of 0.35, yielding a spot size of approxi-
mately 4 μm in diameter. The laser power at the sample
was set to 1 to 2 mW, and the temperature was kept at
77 K, using a Linkam THMS600 freezing microscope
stage. Accumulation times were 120–300 s with up to
30 repetitions.
All RR spectra were obtained from single MBH crys-
tals. In the standard configuration (θ=0), the long axis
of the needle-shaped MBH crystals, referring to the c-axis
of the crystals unit cell, was always aligned parallel to the
electric field vector E
!of the linearly polarized incident
laser beam (see Figure S7). To vary θ, a half-wave plate in
the excitation pathway of the setup was used to rotate the
electric field vector. This ensured that for each θ,
the same spot on the sample surface was probed. The
polarization-dependent sensitivity of the spectrometer
was compensated by a quarter wave plate placed in front
of the entrance slit. All spectra were calibrated against
the 274.0-cm
1
peak of ice present on the protein
crystals.
3|RESULTS AND DISCUSSION
3.1 |Strategy for identifying RR marker
bands of the various Fe–S clusters
Previously, wavelength-dependent studies on MBH crys-
tals and solutions revealed that by use of a 457 nm excita-
tion the corresponding RR spectra of MBH may include
contributions of all Fe–S centers and the [NiFe] active
site in a redox-dependent manner (Figures 1, 2, and
S4).
[17]
Definition and nomenclature of the various redox
states are given in the Supporting Information (Table S1).
In the H
2
-reduced enzyme, the resonance enhancement
of the Fe–S stretching modes is very weak such that the
contribution of the Fe–S clusters to the RR spectra can be
neglected. Instead, the Ni
a
-L state of the active site gives
rise to distinct RR bands in the region between 400 and
700 cm
1
.
[17,18]
By contrast, in the as-isolated, super-
oxidized enzyme, the active site resides predominately in
the Ni
r
-B state, which is characterized by a hydroxy
group bridging the nickel and iron and minor amounts of
the one electron reduced Ni
r
-S state. Active site-related
RR bands of the potential photoproduct of the latter, lac-
king the bridging ligand, the so called Ni
a
-S state, are
hardly detectable.
[18]
Therefore, this spectral region is
dominated by the Fe–OH modes of the superoxidized
proximal cluster (Figure 2).
[12,18]
The proximal cluster in
both superoxidized states, with and without the Fe-bound
hydroxy ligand, as well as the oxidized medial and distal
clusters give rise to relatively strong RR bands between
300 and 400 cm
1
(Figure 2). Thus, the spectrum may
include contributions from up to four oxidized Fe–S clus-
ter states (Table 1). To identify the main Fe–S modes of
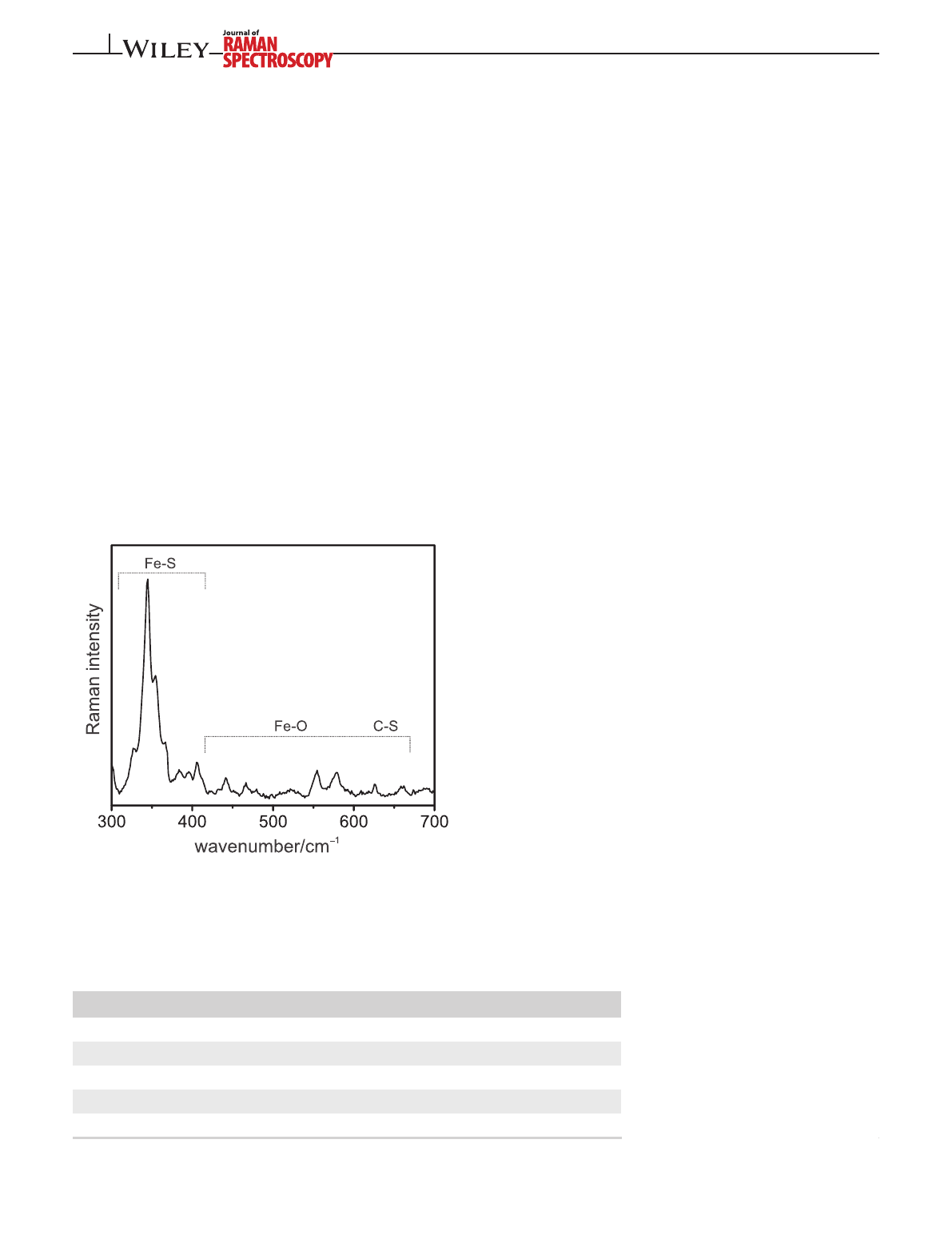
FIGURE 2 Resonance Raman (RR) spectrum of a single
crystal of superoxidized membrane-bound [NiFe]-hydrogenase
(MBH), showing the regions of the Fe–S stretching and Fe–OH/C–S
modes. The spectrum was obtained with 457-nm excitation at 77 K
TABLE 1 Fe–S cluster types
present at the proximal, medial and
distal positions of the electron transfer
chains in MBH proteins used for RR
spectroscopic analysis
Variant/redox state Proximal Medial Distal
MBH
native
/superoxidized OH-occupied [4Fe–3S] [3Fe–4S] [4Fe–4S]
MBH
native
/superoxidized OH-free [4Fe–3S] [3Fe–4S] [4Fe–4S]
MBH
native
/ascorbate-reduced [4Fe–3S]
a
[3Fe–4S] [4Fe–4S]
MBH
P242C
, superoxidized OH-occupied [4Fe–3S] [4Fe–4S] [4Fe–4S]
MBH
C19G/C120G
, oxidized [4Fe–4S] [3Fe–4S] [4Fe–4S]
Abbreviations: MBH, membrane-bound [NiFe]-hydrogenase; RR, Resonance Raman.
a
(partially) Reduced state; not detectable in the RR spectrum.
2624 SIEBERT ET AL.

the individual Fe–S cluster states, we employed different
approaches to simplify the spectra via spectral discrimi-
nation. These were (i) orientation-dependent RR mea-
surements of MBH crystals to achieve a preferential
enhancement of the Raman bands of individual clusters,
(ii) selective (partial) reduction of the proximal cluster by
ascorbate, and (iii) conversion of the proximal [4Fe–3S]
and medial [3Fe–4S] clusters into [4Fe–4S] cluster species
by site-directed amino acid exchanges (Table 1).
3.2 |Orientation-dependent RR
spectroscopy of MBH crystals
As shown in earlier studies, the most prominent RR
bands of the [4Fe–4S] and [3Fe–4S] clusters were
observed at approximately 337 and 348 cm
1
, respec-
tively, which are attributed to the same type of mode
(ν
b
).
[19,20]
It originates from the Fe–S stretchings includ-
ing bridging sulfur atoms. The excitation profile and the
low depolarization ratio of the mode point to a totally
symmetric character with reference to an ideal cube-
derived symmetry. Its intensity is mainly provided via an
A-term enhancement mechanism in resonance with the
electronic charge-transfer transition. Correspondingly,
one would expect that the intensity scales with (cosβ)
2
,
where βis the angle defined by the vectors of the electric
field E
!and the transition dipole moment μ
!.
[21]
Given
that the individual transition dipole moments of the three
Fe–S clusters adopt different orientations in the protein,
there will be a specific angle β, which ensures a selective
enhancement of the modes of one cluster compared with
those of the other two clusters. To exploit this principle
for an efficient spectral discrimination in orientation-
dependent RR spectroscopy of single protein crystals, an
identical orientation of all molecules in the unit cell is
required. However, MBH crystallizes in the orthorhombic
space group P2
1
2
1
2
1
, including four asymmetric units,
each of them harboring one MBH heterodimer.
[14]
Thus,
the projection of μ
!of a given Fe–S center on the c-axis of
the crystal and, thus, on E
!is different for each hydroge-
nase molecule in the unit cell. Consequently, the angle
dependence of the cluster-specific enhancement of ν
b
is
somewhat blurred. This is also true for the second main
band located between 360 and 368 cm
1
, which is related
to Fe–S stretchings involving the terminal cysteine sulfur
atoms (ν
t
).
[10,20]
Because this mode is most likely
enhanced via vibronic coupling (B-term scattering), the
intrinsic enhancement does not follow a simple (cosβ)
2
dependence. As a consequence, the angle-dependent RR
spectra of MBH single crystals are expected to include
varying contributions of the individual clusters but do
not display a complete spectral separation.
For the orientation-dependent experiments, aerobi-
cally grown MBH crystals were used. Among the various
crystals prepared in this way, the ratio between the
superoxidized state with and without hydroxyl ligand
varied substantially.
[12]
For the present angle-dependent
measurements, we therefore selected crystals with the
highest possible OH ligand population of the [4Fe–3S]
cluster as determined by the characteristic Fe–OH modes
between 500 and 600 cm
1
(vide infra).
[12,17]
The
orientation-dependent spectra were obtained from such a
single MBH crystal, with increments of typically 20in
the range from θ=0to 180(Figure S5). Selected exam-
ples are shown in Figure 3. The spectra were normalized
with respect to the integral intensity of the most intense
bands in the region of the Fe–S stretching modes between
330 and 375 cm
1
.
At an angle of 0, the spectrum shows the characteris-
tic signature of the proximal [4Fe–3S] cluster with bound
hydroxyl group in the region above 400 cm
1
, including
the marker bands for the Fe–OH stretching modes at
554 and 578 cm
1
,Fe–O torsional modes below
500 cm
1
, and C–S stretching modes of Cys19 at 625 and
660 cm
1
assigned in our previous work (Figure 3,
right).
[17]
All these bands have maximum relative inten-
sity at parallel excitation (θ=0) such that the
corresponding Fe–S stretching modes are attributed to
band components with peak maxima at 344.5 and
354.5 cm
1
(Figure 3, left). At excitation angles above
50, the spectral pattern changed substantially. The
intensities of the [4Fe–3S] cluster-related bands (with
bound OH) at higher wavenumbers strongly decreased
and became comparable with the very weak bands of the
Ni
a
–S state of the [NiFe] active site (560 and
590 cm
1
),
[17]
which are slightly above the noise level at
90(Figure 3, right). Hence, the spectral changes in the
Fe–S stretching region (Figure 3, left) observed at 70and
90reflect the increased spectral contribution of the
medial and distal Fe–S clusters. These changes include a
broadening of the prominent band envelope at
344.5 cm
1
. The increased intensity on its low-
wavenumber tail points to a stronger enhancement of a
component below 338 cm
1
(Figure 3, left). Furthermore,
the spectrum displays a significant intensity increase of
the 367 cm
1
peak at 70and even more pronounced at
90. This is presumably due to the enhancement of two
closely spaced bands, as concluded from the non-
Lorentzian band profile. For a better illustration of these
changes, we have generated a difference spectrum by
subtracting the spectrum measured at 90from that at 0.
The negative peaks at approximately 336 and 362 cm
1
are in good agreement with those reported for [4Fe–4S]
centers
[19]
and are therefore tentatively attributed to the
distal [4Fe–4S] cluster of MBH.
SIEBERT ET AL.2625
Loading more pages...