
This journal is © The Royal Society of Chemistry 2022 Chem. Commun., 2022, 58, 973–976 | 973
Cite this: Chem. Commun., 2022,
58, 973
Metal-free transfer hydrochlorination of internal
C–C triple bonds with a bicyclo[3.1.0]hexane-
based surrogate releasing two molecules of
hydrogen chloride†
Andreas J. Weidkamp and Martin Oestreich *
The development and application of a transfer hydrochlorination
reagent based on a trichlorinated bicyclo[3.1.0]hexane core that
transfers two molecules of HCl per molecule of surrogate to a p-
basic substrate under B(C
6
F
5
)
3
catalysis is reported. Lewis acid-
assisted chloride abstraction followed by thermal electrocyclic
cyclopropyl-to-allyl cation ring opening releases ring strain as a
previously unexploited driving force.
Alkenyl chlorides are important building blocks in organic
chemistry both on industrial and laboratory scale. For their
synthesis, the addition of HCl across C–C triple bonds is
arguably the most atom-economic way of forming the C(sp
2
)–
Cl bond. However, this direct approach requires the use of
corrosive and highly toxic HCl gas. Moreover, in the industrial
benchmark process of hydrochlorination of acetylene to gen-
erate vinyl chloride, less appealing mercury-based catalysts are
the state of the art although newer protocols using expensive
palladium-
1
or gold-based
2
catalysts have been developed. In
more complex molecules, uncatalyzed, direct hydrochlorina-
tion with HCl is usually limited to electron-rich terminal
alkynes,
3
while ruthenium-,
4
palladium-,
5
platinum-,
6
and
gold-based
7
catalysts have been shown to also convert a wider
range of substrates including electron-deficient alkynes.
8,9
Uti-
lizing readily activated surrogates of commonly employed
reagents can both solve handling problems and allow for the
use of cheaper and less toxic catalysts albeit at the expense of
reduced atom efficiency. Surrogates that contain more than one
equivalent of the compound to transfer could be even more
attractive but are currently not established.
A broadly applicable surrogate with reduced acidity com-
pared to HCl was introduced by Hammond and Xu.
10
Although
a gold catalyst was typically required, a handful of more
activated substrates reacted under metal-free conditions. Based
on their research interest in transition-metal-catalyzed shuttle
catalysis,
11
Morandi and coworkers established non-acidic
hydrochlorination surrogates.
12
In a first protocol, 4-
chlorobutan-2-one was used as surrogate, yielding highly reac-
tive methyl vinyl ketone as stoichiometric byproduct. While
iridium catalysis was required, high functional-group tolerance
was observed. Under more forcing conditions, the second
protocol enabled the use of cheap tert-butyl chloride and
liberated less reactive isobutene yet at the cost of a limited
substrate scope.
A general strategy for ionic transfer hydrofunctionalizations
employed by our group relies on the use of strong Brønsted or
Lewis acids such as B(C
5
F
6
)
3
to activate otherwise bench-stable
surrogates.
13
Typically, these processes employ pro-aromatic
reagents Ipassing through a reactive Wheland intermediate II
before affording an aromatic byproduct III (Scheme 1, top).
13,14
In this vein, we have successfully developed protocols for
Scheme 1 Established hydrofunctionalization surrogates and develop-
ment of a new surrogate for metal-free transfer hydrochlorination.
Institut fu
¨r Chemie, Technische Universita
¨t Berlin, Strasse des 17. Juni 115,
10623 Berlin, Germany. E-mail: martin.oestreich@tu-berlin.de
†Electronic supplementary information (ESI) available: Experimental details and
characterization data. See DOI: 10.1039/d1cc06591b
Received 23rd November 2021,
Accepted 17th December 2021
DOI: 10.1039/d1cc06591b
rsc.li/chemcomm
ChemComm
COMMUNICATION
Open Access Article. Published on 20 December 2021. Downloaded on 4/11/2022 7:30:44 AM.
This article is licensed under a
Creative Commons Attribution 3.0 Unported Licence.
View Article Online
View Journal
| View Issue

974 | Chem. Commun., 2022, 58, 973–976 This journal is © The Royal Society of Chemistry 2022
transfer hydro(pseudo)halogenation such as transfer hydro-
cyanation,
15
-iodination
16
and -bromination
17
using surrogates
that show no Brønsted acidity and do not possess protic
hydrogen atoms (Scheme 1, middle). While for transfer hydro-
cyanation a simple 3-substituted cyclohexa-1,4-diene scaffold
could be utilized, this strategy had to be refined for the transfer
of HI and HBr. In these cases, the corresponding 3-
halocyclohexa-1,4-dienes were not suitable as surrogates due
to their chemical instability at room temperature. The solution
that enabled the transfer of HI was the installation of an
ethylene tether to stabilize the molecule that would eventually
be cleaved to generate ethylene gas and the aforementioned
Wheland intermediate. When adapting this concept to transfer
hydrobromination, we realized that activation of the surrogate
was the easier the weaker the C(sp
3
)–X bond with X = I and Br as
well as Cl. Overcoming this difficulty required higher tempera-
tures and better stabilization of the intermediate carbenium
ion with an additional phenyl group in the case of hydrobro-
mination. These findings indicated that a transfer hydrohalo-
genation becomes increasingly complicated for the lighter
halogen homologues and that a more sophisticated surrogate
design might be necessary.
We commenced our quest for an HCl surrogate based on our
experiences outlined above. Simple cyclohexa-1,4-diene surro-
gates were deemed as too unstable and, after preliminary
experiments, we also learned that cleavage of the relatively
strong C(sp
3
)–Cl bond in ethylene-tethered surrogates was
impossible at synthetically useful temperatures.
18
We then
devised the platform 1for releasing the cationic key intermedi-
ate based on the thermal, disrotatory electrocyclic ring opening
of bicyclo[3.1.0]hexanes (Scheme 1, bottom).
19–21
After chloride
abstraction, we intended to harness release of ring strain as a
driving force for the generation of the desired Wheland
complex 2. Subsequent aromatization to byproduct 3would
transfer the proton to the substrate. The parent system 1is,
however, described in the literature as thermally labile.
19
For
this reason, we considered and prepared the related surrogates
6–9 (Scheme 2, bottom) as potential alternatives.
Benzannulated surrogate 6was obtained in one step from
indene as a white solid in 40% yield by dichlorocarbene
addition.
22
When employed in a test reaction with 1-
phenylprop-1-yne (4a) at 100 1CinC
6
D
6
with 10 mol% of
B(C
6
F
5
)
3
as catalyst, we found the formation of the desired
vinyl chloride 5a in 97% NMR yield after 19 h (Scheme 2, top).
Encouraged by this initial result, we envisioned that the double
bond present in 1could also be generated during the transfer
reaction by another elimination of HCl. A potential surrogate
would thus carry two molecules of HCl, thereby allowing for the
transfer of both. Surrogate cis-7bearing an additional chloro-
substituent on the cyclopentane ring was prepared and tested
in the model reaction.
23
Surprisingly, no product formation
could be detected even after prolonged reaction time. The
independently tested diastereomer trans-7was equally unreac-
tive, indicating that it is not steric hindrance imposed by that
chlorine atom that inhibits the reaction. We hypothesized that
the activation of the surrogate is facilitated by better
stabilization of the carbenium ion intermediate. We therefore
prepared the phenyl-substituted surrogate 8, available in seven
steps from furfuryl alcohol in 1.2% overall yield. Indeed, this
surrogate successfully transferred two molecules of HCl. The
reaction time was significantly longer than that seen with
surrogate 6. We attribute this to differences in orbital overlap
of the aromatic system with the carbenium ion. On the one
hand, the rigid geometry of 6enforces a planar benzyl cation.
Orbitals are then perfectly aligned for carbenium ion stabili-
zation by the phenyl ring and the adjacent double bond. On the
other hand, the phenyl ring in the cation derived from 8can
twist out of plane to avoid steric hindrance. This renders a
system that rather resembles a mere allyl cation. In contrast,
previous transfer-hydrobromination surrogates benefitted from
installation of a phenyl group.
17
As a general disadvantage of
the productive surrogates 6and 8, the byproducts 2-
chloronaphthalene and -biphenyl are difficult to remove, and
the latter is assumed to be highly toxic.
To provide carbeniumion stabilization without introducing
a ‘‘heavy’’ phenyl group, we considered two effects: (1) a methyl
group at the bridgehead position should lend further stabili-
zation and (2) an adequately positioned chlorine atom could
allow for the formation of a chloronium ion.
24
We found that
we could test the influence of both effects with the trichlor-
oalkane trans-9. The requisite cis-configured alcohol for its
synthesis was described in the literature as a rare example
where a hydroxy function acts as a directing group for dichlor-
ocarbene addition.
25
Whereas any of the previous syntheses did
Scheme 2 Developed surrogates and their properties.
a
Volatile was
arbitrarily defined as having a boiling point below 160 1C.
b
Yield and d.r.
obtained in the model reaction were determined by
1
H NMR spectroscopy
with dimethyl terephthalate as an internal standard.
c
Reactions at 100 1C
and 140 1C were run in C
6
D
6
and C
6
D
5
Cl, respectively.
Communication ChemComm
Open Access Article. Published on 20 December 2021. Downloaded on 4/11/2022 7:30:44 AM.
This article is licensed under a
Creative Commons Attribution 3.0 Unported Licence.
View Article Online
This journal is © The Royal Society of Chemistry 2022 Chem. Commun., 2022, 58, 973–976 | 975
not tolerate unprotected alcohols, a straightforward three-step
synthesis was possible in this case (see the ESI†for details).
Having established the synthetic access, we investigated its
reactivity in our model transfer hydrochlorination. Importantly,
even 0.65 equivalents of the surrogate allowed for a yield of 76%
of 5a after 7 h at 140 1C. This confirmed the planned transfer of
two molecules of HCl from one molecule of the surrogate. The
lower yield compared to that obtained with surrogate 6is
counterbalanced by the unprecedented two-fold transfer and
the generation of a harmless, volatile byproduct. As before, we
were curious about the influence of the surrogate’s relative
stereochemistry and prepared cis-9. In the model reaction, we
observed substantially lower reaction rate, and the product was
obtained in lower yield (51% vs. 76%). This could be due to
increased steric hindrance around the chlorine atoms in cis-9
compared to the situation in trans-9. For optimal transfer
efficiency, diastereopure trans-9was therefore used in all fol-
lowing experiments.
We next turned to the optimization of the reaction para-
meters. The need for an unreactive, high-boiling, and polar
solvent left little room for variation. With the surrogate and the
reaction temperature largely set, we thus focused on testing
different catalysts in chlorobenzene at 130 1C (Table 1). The
weak boron Lewis acid BPh
3
failed to promote the reaction
(entry 2). B(C
6
Cl
5
)
3
, considered a stronger Lewis acid than
B(C
6
F
5
)
3
, was also ineffective, probably due to the increased
steric hindrance around the boron center (entry 3). The
Brønsted acid Tf
2
NH, successfully applied to hydrobromination
and -iodination,
16,17
led to decomposition (entry 4). This find-
ing points to the role of B(C
6
F
5
)
3
as a catalyst instead of being a
simple initiator.
26
As expected, no conversion of the substrate
was detected in the absence of catalyst (entry 5). An increased
catalyst loading of B(C
6
F
5
)
3
accelerated the reaction but was
detrimental to the yield (entry 6 vs. entry 1). Increasing the
reaction temperature from 130 1C to 140 1C drastically reduced
the reaction time and further improved the yield to 76%
(entry 7 vs. entry 1).
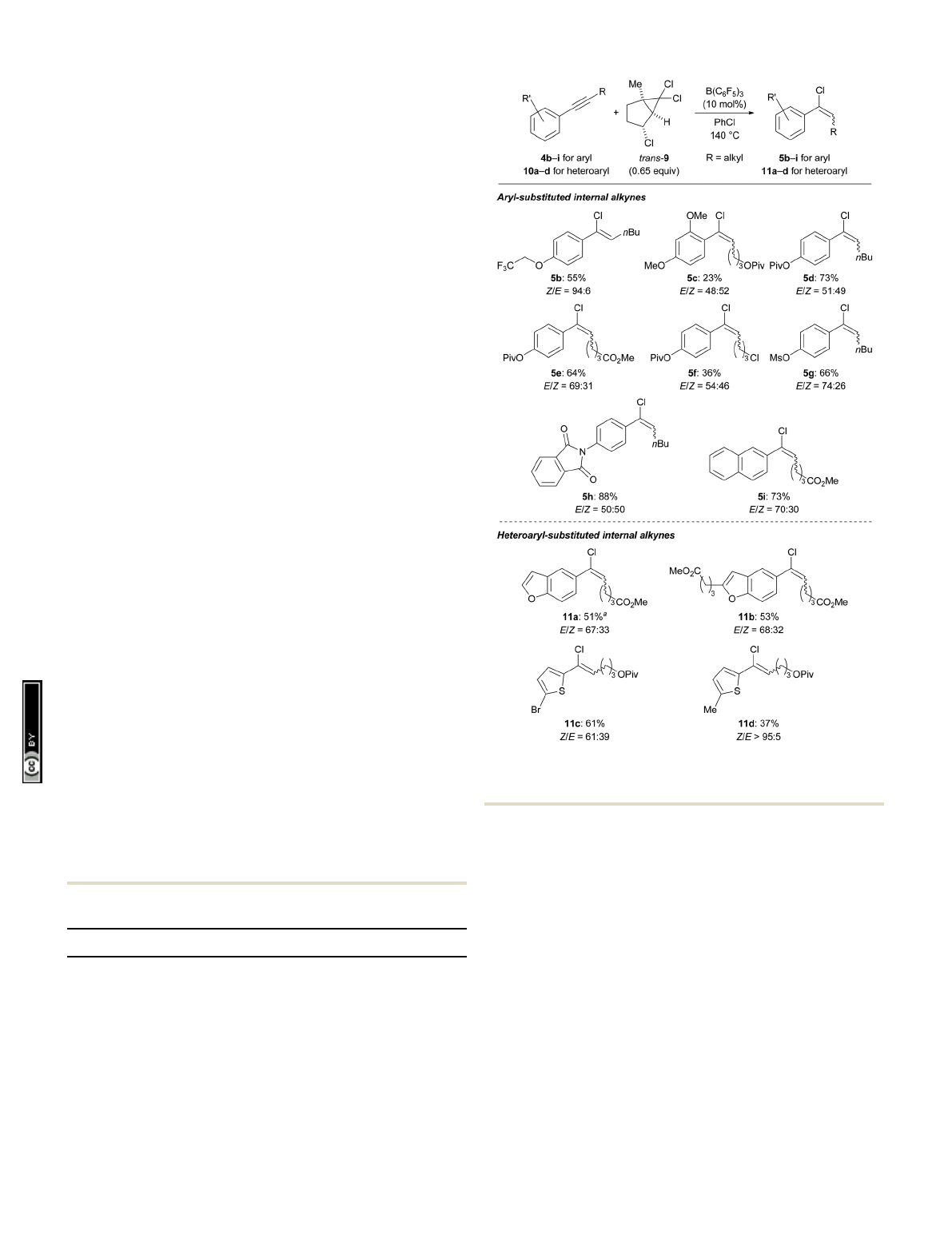
We then set out to explore the substrate scope (Scheme 3,
top). Although initially optimized for the hydrochlorination of
unfunctionalized 1-phenylprop-1-yne (4a), we quickly found
that polar functional groups are tolerated. Their presence also
made the chromatographic separation of the alkenyl chlorides
from generated side products easier. As expected, aryl-
substituted internal alkynes not having electron-withdrawing
groups at the aryl ring participated in this ionic process.
Various phenol derivatives 4b–g either as an ether, ester or
sulfonate reacted in good yields; the dimethoxyaryl group in 4c
is likely too electron-rich for the acidic reaction medium.
Substrate 4e containing two carboxyl groups was also amen-
able. A primary alkyl chloride as in 4f resulted in significant
decomposition. A phthalimide group as in 4h was compatible,
furnishing the desired vinyl chloride in high yield. The naphth-
2-yl-substituted alkyne 4i was converted into the product in
good yield, while a similar substrate having a naphth-1-yl group
was found to decompose under the reaction conditions (see the
ESI†for details).
Table 1 Selected examples of the optimization of the transfer hydro-
chlorination of an internal C–C triple bond (4a -5a;cf. Scheme 2, top)
a
Entry Catalyst (mol%) Time (h) d.r.
b
(E:Z) Yield
b
(%)
1 B(C
6
F
5
)
3
(10 mol%) 83 74 : 26 72
2 BPh
3
(10 mol%) — — o5 (Conv)
3 B(C
6
Cl
5
)
3
(10 mol%) — — o5 (Conv)
4Tf
2
NH (10 mol%) — — Decomp
5 None — — o5 (Conv)
6 B(C
6
F
5
)
3
(20 mol%) 2 74 : 26 65
7
c
B(C
6
F
5
)
3
(10 mol%) 7 74 : 26 76
a
Unless otherwise noted, all reactions were performed in C
6
D
5
Cl at
130 1C using surrogate trans-9(0.65 equiv.).
b
Diastereomeric ratio,
yield, and conversion were determined by
1
H NMR spectroscopy with
dimethyl terephthalate as an internal standard.
c
Reaction performed
at 140 1C.
Scheme 3 Substrate scope of aryl/heteroaryl-substituted internal alkynes
suitable for the transfer hydrochlorination.
a
74% (E/Z= 71 : 29) were
obtained on a 2.0 mmol scale.
ChemComm Communication
Open Access Article. Published on 20 December 2021. Downloaded on 4/11/2022 7:30:44 AM.
This article is licensed under a
Creative Commons Attribution 3.0 Unported Licence.
View Article Online
976 | Chem. Commun., 2022, 58, 973–976 This journal is © The Royal Society of Chemistry 2022
We next investigated a variety of heteroaryl groups, poten-
tially not compatible with B(C
6
F
5
)
3
at high temperature
(Scheme 3, bottom). Notably, benzofurans turned out to be
suitable substrates as exemplified by 10a and the 2-substituted
derivative 10b. The reaction of a furyl-substituted derivative was
successful but gave an unstable product that could not be
isolated (see the ESI†for details). Thiophene derivatives such
as 10c and 10d underwent the hydrochlorination without any
problems.
The stereoselectivity of the reaction was generally modest,
with the E/Zratio ranging from 74 : 26 to 39 : 61 in most cases.
The obtained major isomer was normally E-configured which is
in accordance with a vinyl cation as an intermediate accepting a
chloride anion from the less hindered side either from the
in situ-formed borate [ClB(C
6
F
5
)
3
]
or the surrogate itself.
27
The present work is conceptual rather than being an
advance in synthetic methodology. We have described a design
for storable surrogates of HCl as an alternative to unstable or
unreactive cyclohexa-1,4-diene-based platforms. Aromatization
of the multiply chlorinated bicyclo[3.1.0]hexanes can be
initiated by B(C
6
F
5
)
3
-mediated chloride abstraction from a
geminally dichlorinated cyclopropane unit. An electrocyclic
ring opening driven by the release of strain followed by loss
of a proton leads to a chlorobenzene byproduct with low
molecular weight. The surrogate can be designed in such a
way that another elimination of HCl is required to achieve
aromatization. By this, two molecules of HCl can be generated
from one molecule of the surrogate. With an internal C–C triple
bond as a proton acceptor,
28
a B(C
6
F
5
)
3
-catalyzed transfer
hydrochlorination has become possible in moderate yields.
A. J. W. gratefully acknowledges the Berlin Graduate School
of Natural Sciences and Engineering for a predoctoral fellow-
ship (2017–2020). M. O. is indebted to the Einstein Foundation
Berlin for an endowed professorship. We thank Markus Budde
for his experimental contributions as well as Dr Weiqiang Chen
and Pedro Helou de Oliveira for helpful discussions (all TU
Berlin).
Conflicts of interest
There are no conflicts to declare.
Notes and references
1 J. Zhao, Y. Yue, G. Sheng, B. Wang, H. Lai, S. Di, Y. Zhai, L. Guo and
X. Li, Chem. Eng. J., 2019, 360, 38–46.
2 J. Zhao, B. Wang, X. Xu, Y. Yu, S. Di, H. Xu, Y. Zhai, H. He, L. Guo,
Z. Pan and X. Li, J. Catal., 2017, 350, 149–158.
3(a) P. J. Kropp and S. D. Crawford, J. Org. Chem., 1994, 59,
3102–3112; (b) C.-X. Xu, C.-H. Ma, F.-R. Xiao, H.-W. Chen and
B. Dai, Chin. Chem. Lett., 2016, 27, 1683–1685.
4(a) H. Klein, T. Roisnel, C. Bruneau and S. De
´rien, Chem. Commun.,
2012, 48, 11032–11034; (b)S.De
´rien, H. Klein and C. Bruneau,
Angew. Chem., Int. Ed., 2015, 54, 12112–12115.
5(a) J. Dupont, N. R. Basso, M. R. Meneghetti, R. A. Konrath,
R. Burrow and M. Horner, Organometallics, 1997, 16, 2386–2391;
(b) G. Zhu, D. Chen, Y. Wang and R. Zheng, Chem. Commun., 2012,
48, 5796–5798; (c) J. Derosa, A. L. Cantu, M. N. Boulous,
M. L. O’Duill, J. L. Turnbull, Z. Liu, D. M. de La Torre and
K. M. Engle, J. Am. Chem. Soc., 2017, 139, 5183–5193;
(d) D. A. Petrone, I. Franzoni, J. Ye, J. F. Rodrı
´guez, A. I. Poblador-
Bahamonde and M. Lautens, J. Am. Chem. Soc., 2017, 139,
3546–3557.
6 C.-Y. Lo, M. P. Kumar, H.-K. Chang, S.-F. Lush and R.-S. Liu, J. Org.
Chem., 2005, 70, 10482–10487.
7 J. Oliver-Meseguer, A. Dome
´nech-Carbo
´, M. Boronat, A. Leyva-Pe
´rez
and A. Corma, Angew. Chem., Int. Ed., 2017, 56, 6435–6439.
8 For a review on transition-metal-catalyzed carbon–halogen bond
formation, see: D. A. Petrone, J. Ye and M. Lautens, Chem. Rev.,
2016, 116,8003–8104.
9 The uncatalyzed, Michael-type addition of HCl to electron-deficient
alkynes is long known. See: (a) R. Friedrich, Justus Liebigs Ann.
Chem., 1883, 219, 368–374; (b) A. Michael and H. Pendleton, J. Prakt.
Chem., 1889, 40, 63–68.
10 (a) R. Ebule, S. Liang, G. B. Hammond and B. Xu, ACS Catal., 2017,
7, 6798–6801; (b) X. Zeng, Z. Lu, S. Liu, G. B. Hammond and B. Xu,
J. Org. Chem., 2017, 82, 13179–13187; (c) S. Liang, G. B. Hammond
and B. Xu, Green Chem., 2018, 20, 680–684; (d) X. Zeng, S. Liu,
G. B. Hammond and B. Xu, ACS Catal., 2018, 8, 904–909.
11 For general aspects of shuttle catalysis, see: (a) B. N. Bhawal and
B. Morandi, Angew. Chem., Int. Ed., 2019, 58, 10074–10103; for an
aromatization-driven transfer dichlorination, see: (b) X. Dong,
J. L. Roeckl, S. R. Waldvogel and B. Morandi, Science, 2021, 371,
507–514.
12 P. Yu, A. Bismuto and B. Morandi, Angew. Chem., Int. Ed., 2020, 59,
2904–2910.
13 (a) J. C. L. Walker and M. Oestreich, Synlett, 2019, 30, 2216–2232;
(b) S. Keess and M. Oestreich, Chem. Sci., 2017, 8, 4688–4695.
14 For radical-based approaches, see: A. Bhunia and A. Studer, Chem,
2021, 7, 2060–2100.
15 P. Orecchia, W. Yuan and M. Oestreich, Angew. Chem., Int. Ed., 2019,
58, 3579–3583.
16 W. Chen, J. C. L. Walker and M. Oestreich, J. Am. Chem. Soc., 2019,
141, 1135–1140.
17 W. Chen and M. Oestreich, Org. Lett., 2019, 21,4531–4534.
18 H. Fang and M. Oestreich, Unpublished results, Technische Univer-
sita
¨t, Berlin, 2019.
19 M. S. Baird, D. G. Lindsay and C. B. Reese, J. Chem. Soc. C, 1969,
1173–1178.
20 For the electrocyclic ring opening of cyclopropyl cations, see:
(a) R. B. Woodward and R. Hoffmann, J. Am. Chem. Soc., 1965, 87,
395–397; (b) P. v. R. Schleyer, T. M. Su, M. Saunders and
J. C. Rosenfeld, J. Am. Chem. Soc., 1969, 91, 5174–5176; Orbital
symmetry dictates that in the present system, the chloride ion is
released from the axial (= ‘‘endo’’) position. This has been experi-
mentally verified: (c) P. S. Skell and S. R. Sandler, J. Am. Chem. Soc.,
1958, 80, 2024–2025; (d) M. S. Baird and C. B. Reese, Tetrahedron
Lett., 1967, 8, 1379–1382.
21 For a similar approach exploiting the ring strain of pro-aromatic
bicyclo[2.2.0]hexenes, see: B. Wu, J. Wang, X. Liu and R. Zhu, Nat.
Commun., 2021, 12, 3680.
22 (a) W. E. Parham and H. E. Reiff, J. Am. Chem. Soc., 1955, 77,
1177–1178; (b) W. E. Parham, H. E. Reiff and P. Swartzentruber,
J. Am. Chem. Soc., 1956, 78, 1437–1440; for a seminal report, see:
(c) G. L. Ciamician and M. Dennstedt, Ber. Dtsch. Chem. Ges., 1881,
14, 1153–1163.
23 I. Fleming and E. J. Thomas, Tetrahedron, 1972, 28, 4989–5001.
24 (a) G. A. Olah and J. M. Bollinger, J. Am. Chem. Soc., 1967, 89,
4744–4752; (b) G. A. Olah, J. M. Bollinger and J. Brinich, J. Am. Chem.
Soc., 1968, 90, 2587–2594.
25 (a) D. Seyferth and V. A. Mai, J. Am. Chem. Soc., 1970, 92, 7412–7424;
(b) R. H. Ellison, J. Org. Chem., 1980, 45, 2509–2511.
26 S. Banerjee and K. Vanka, ACS Catal., 2018, 8, 6163–6176.
27 As exceptions, the electron-rich substrates 4b and 10d gave almost
pure Z-isomers as products. Presumably, thermodynamic control
favors isomerization to the Zisomer. As a general trend, the
stereoselectivity of the reaction was found to considerably vary with
the alkyne substrate but to be almost independent of the used
surrogate. This indicates that chloride is released from the surro-
gate before the C(sp
2
)–Cl bond is formed. As an alternative explana-
tion for the observed diastereomeric ratios, thermodynamic
equilibration of the alkenyl chlorides cannot be ruled out.
28 Terminal alkynes decomposed and dialkyl-substituted internal
alkynes did not react.
Communication ChemComm
Open Access Article. Published on 20 December 2021. Downloaded on 4/11/2022 7:30:44 AM.
This article is licensed under a
Creative Commons Attribution 3.0 Unported Licence.
View Article Online
Loading more pages...