German Edition:DOI:10.1002/ange.201508879
Synthetic Methods International Edition:DOI:10.1002/anie.201508879
Transfer Hydrosilylation
Martin Oestreich*
Dedicated to Professor Siegfried Ble-
chert on the occasion of his 70th birthday
cations ·cyclohexa-1,4-dienes ·hydrosilylation ·radi-
cals ·silane transfer
1. Concept and Strategy
Anaive approach to understanding the chemistry of
dihydrogen is to look at hydrosilanes.Anextreme view is that
the silicon atom is nothing but a“fat” hydrogen atom.
Although this is clearly agross oversimplification, the Si¢H
bond often serves as auseful model of the stronger H¢H
bond, particularly in cases where both participate in the same
net reaction.
Acore area of the dihydrogen arena without acounterpart
in silicon chemistry is transfer hydrogenation. Known for
more than acentury,both heterogeneous and homogeneous
methods have found broad application in industrial and
academic settings.[1] Conversely,conceptually related transfer
hydrosilylation processes had remained largely unknown until
the research groups of Studer and Oestreich independently
disclosed radical and ionic transfer hydrosilylations.Both
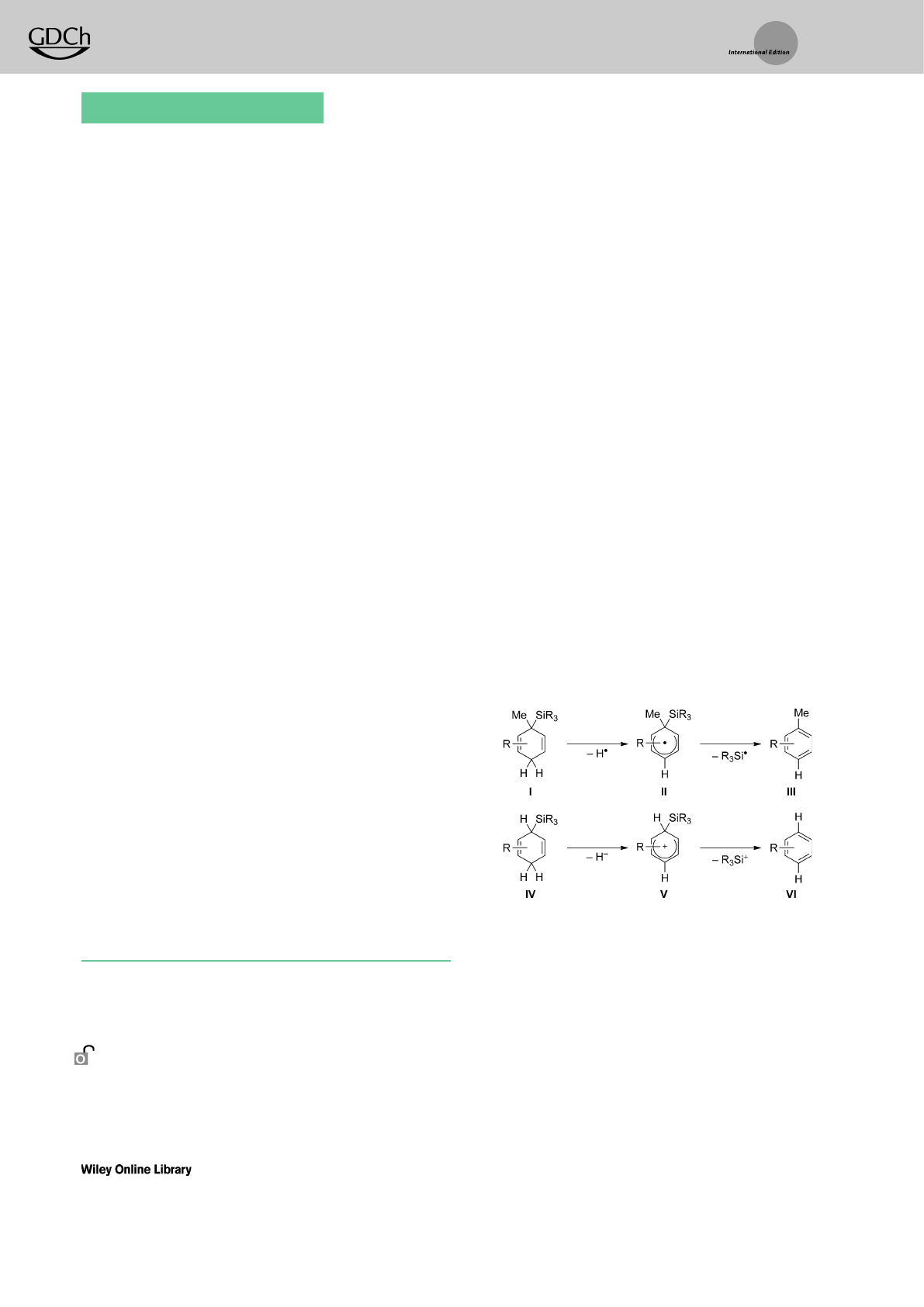
techniques are based on silicon-substituted cyclohexa-1,4-
dienes and exploit aromatization as the driving force
(Scheme 1). Thestepwise release of the hydrosilane provides
equivalents of homolytically (I!II!III,top) or heterolyti-
cally (IV!V!VI,bottom) cleaved Si¢Hbonds.The radical
sequence is initiated by hydrogen atom abstraction from the
methylene group in I(I!II)followed by fragmentation of II
(II!III).[2] Theionic pathway consists of hydride abstraction
from IV to yield the silicon-stabilized cyclohexadienyl cation
V[3] (IV!V)and formal dissociation of the silicon cation
(V!VI). As opposed to the heterolytic cleavage of the Si¢C
bond (V!VI), substitution at the silicon-bearing carbon
atom in cyclohexadienyl radical II is required for selective Si¢
Cbond homolysis (II!III)because otherwise release of
ahydrogen atom competes.[4]
Transfer hydrogenation is without question acommon technology in
industry and academia. Unlike its countless varieties,conceptually
related transfer hydrosilylations had essentially been unreported until
the recent development of aradical and an ionic variant. The new
methods are both based on asilicon-substituted cyclohexa-1,4-diene
and hinge on the aromatization of the corresponding cyclohexadienyl
radical and cation intermediates,respectively,concomitant with homo-
or heterolytic fission of the Si¢Cbond. Both the radical and ionic
transfer hydrosilylation are brought into context with one other in this
Minireview,and early insight into the possibility of transfer hydro-
silylation is included. Although the current state-of-the-art is certainly
still limited, the recent advances have already revealed the promising
potential of transfer hydrosilylation.
Scheme 1. Silicon-substituted cyclohexa-1,4-dienes as hydrosilane sur-
rogates in radical and ionic sequences.
[*] Prof. Dr.M.Oestreich
Institut fír Chemie, Technische Universitt Berlin
Strasse des 17. Juni 115, 10623 Berlin (Germany)
E-mail:m[email protected]
Homepage:http://www.organometallics.tu-berlin.de
Ó2015 The Authors. Published by Wiley-VCH Verlag GmbH &Co.
KGaA. This is an open access article under the terms of the Creative
Commons AttributionNon-CommercialNoDerivs License, which
permitsuse and distribution in any medium, provided the original
work is properly cited, the use is non-commercial and no modifica-
tions or adaptations are made.
A
ngewandte
Chemie
Minireviews
494 Ó2016The Authors. Published by Wiley-VCH Verlag GmbH &Co. KGaA, Weinheim Angew.Chem. Int. Ed. 2016,55,494 –499
Thestrategy outlined above is the basis for the develop-
ment of radical as well as ionic transfer hydrosilylations.This
Minireview discusses and compares both approaches and
highlights experimental findings that have gone almost
unnoticed.
2. Radical Transfer Hydrosilylation
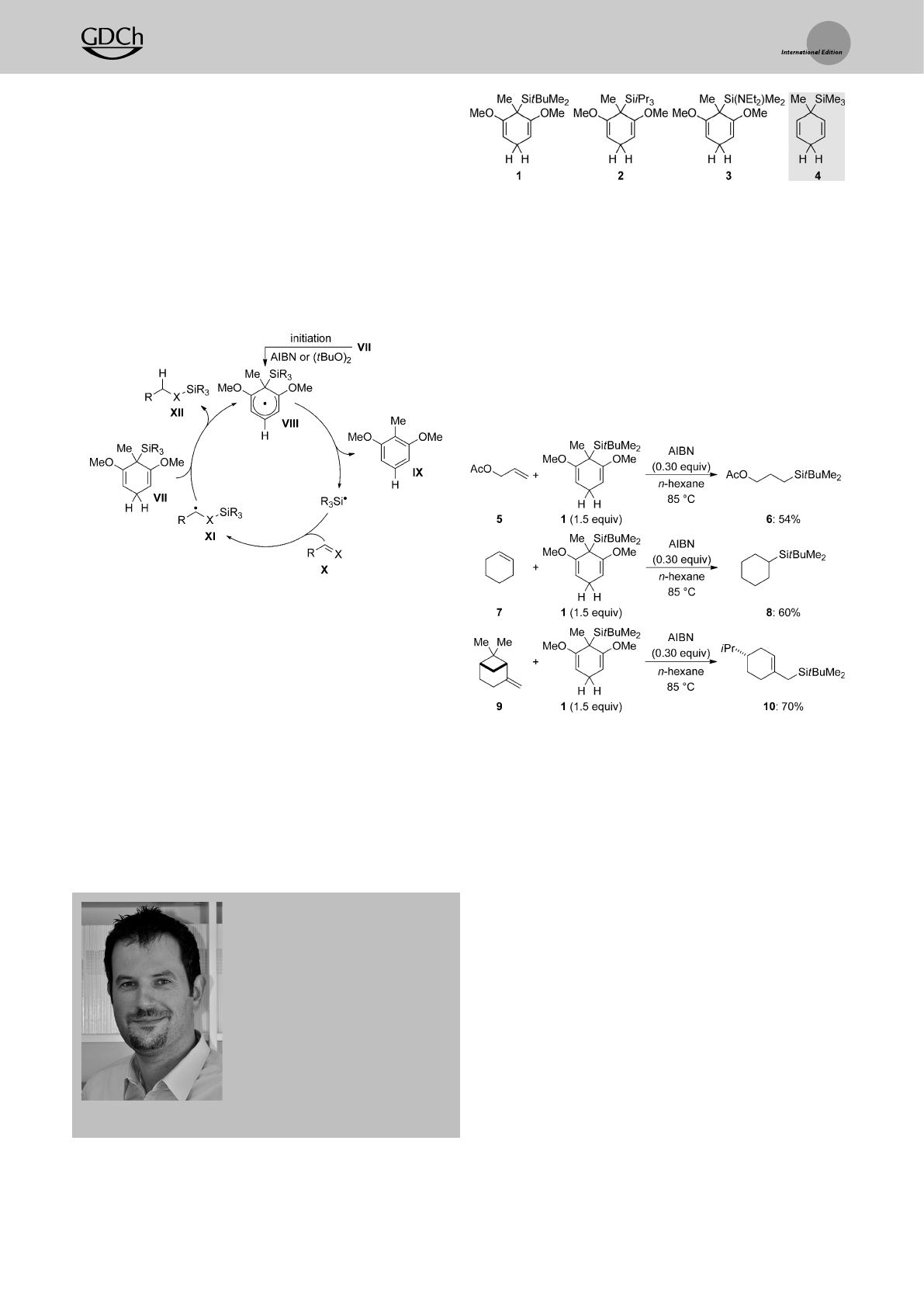
Studer and co-workers introduced and established silicon-
substituted cyclohexa-1,4-dienes of type VII as radical chain
reducing reagents (Scheme 2).[5] As an alternative to conven-
tional tin hydrides,reagents VII enable various reductive
defunctionalizations.[2,5,6] Amrein and Studer also realized
that unsaturated radical acceptors such as Xwould allow the
formal transfer of ahydrosilane from the cyclohexa-1,4-diene
VII to the p-system of X(Scheme 2).[7,8] After initiation, the
cyclohexadienyl radical VIII transfers the silicon fragment to
acceptor X(VII!VIII!IX)toyield b-silicon-substituted
radical XI. XI then acts as the chain carrier,abstracting
ahydrogen atom from VII (VII!VIII)concomitant with
formation of the hydrosilylated acceptor XII (XI!XII).
Representative examples of VII are resorcinol-derived
cyclohexa-1,4-dienes 1–3with trialkylsilyl or heteroatom-
substituted dialkylsilyl groups; 4is an attractive surrogate for
gaseous Me3SiH (Figure 1). As mentioned above,the methyl
group at the silicon-bearing carbon atom in 1–4is necessary to
sustain the chain reaction.[2] Aryl substituents at the silicon
atom are also tolerated (not shown).
Theuse of 1(1.5 equiv) as atransfer reagent is illustrated
for afew selected examples (Scheme 3).[7] Both terminal and
internal alkenes react in decent yields (5!6and 7!8). The
acetate group is compatible with the radical process but will
be too Lewis-basic for the ionic variant (see below). The1,1-
disubstituted double bond in b-pinene also accepts the silicon
radical, but the initially formed radical intermediate under-
goes fast ring opening prior to reduction with 1(9!10). These
transformations are the seminal examples of transfer hydro-
silylation. Thestrength of this process is that it brings about
radical hydrosilylation, which usually fails with trialkylsilanes
as aresult of their relatively strong Si¢Hbonds compared to,
for example,(Me3Si)3SiH.
Radical transfer hydrosilylation coupled with 5-exo-trig
cyclizations is shown for different transfer reagents in
Scheme 4.[7,8] Thelevel of diastereocontrol is generally low,
but the slight preference for the cis relative configuration is
explained with the Beckwith–Houk model for these ring
closures.The transfer of tBuMe2SiH released from 1proceeds
under the previously employed setup (AIBN at 8588C; 11!
12). However,the reaction of atwofold excess of Me3SiH
surrogate 4required harsher reaction conditions [(tBuO)2at
14088C; 11!13)].Itisnoteworthy that even the transfer of
TamaoÏs (Et2N)Me2SiH from 3works in reasonable yield,
thereby providing ahandle for further functional group
manipulations (11!14 after treatment with isopropanol).
Martin Oestreich (born in 1971 in Pforz-
heim/Germany) is Professor of Organic
Chemistry at the Technische Universitt
Berlin. He received his diploma with Paul
Knochel (Marburg, 1996) and his PhD with
Dieter Hoppe (Mínster,1999). After atwo-
year postdoctoral stint with Larry E. Over-
man (Irvine, 1999–2001),hecompleted his
habilitation with Reinhard Bríckner (Frei-
burg, 2001–2005) and was appointed Pro-
fessor of Organic Chemistry at the
Westflische Wilhelms-Universitt Mínster
(2006–2011). He also held visiting positions
at Cardiff University in Wales (2005) and at The Australian National
University in Canberra (2010).
Scheme 2. Radical chain of the radical transfer hydrosilylation. X=CH2
(alkenes) and O(aldehydes). AIBN=azobisisobutyronitrile.
Figure 1. Typical silicon-substituted cyclohexa-1,4-dienes for radical
transfer hydrosilylation.
Scheme 3. Radical transfer hydrosilylation of alkenes.
A
ngewandte
Chemie
Minireviews
495Angew.Chem. Int.Ed. 2016,55,494 –499 Ó2016 The Authors. Published by Wiley-VCH Verlag GmbH &Co. KGaA, Weinheim www.angewandte.org
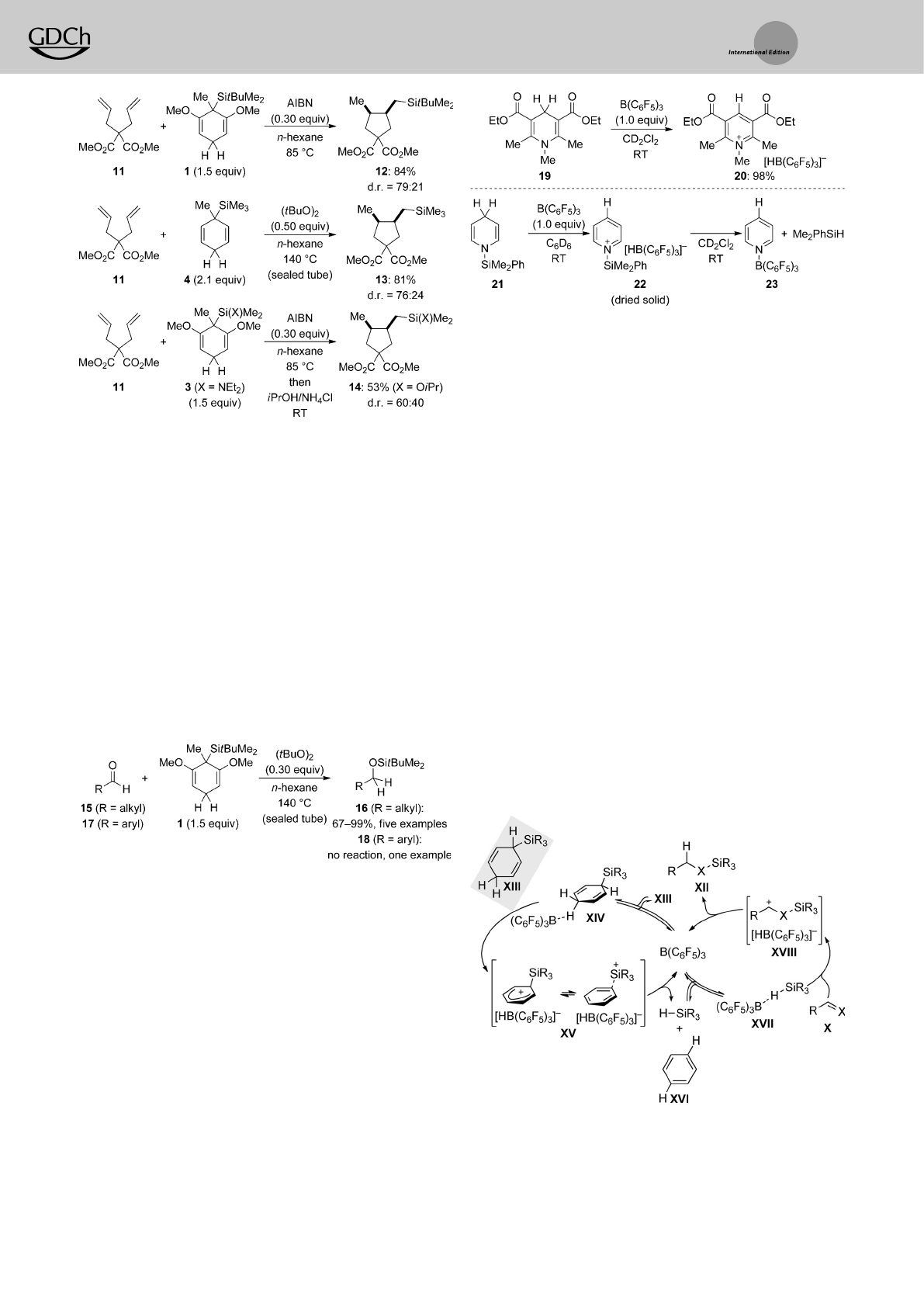
Selected triple bonds also participated in the radical transfer
hydrosilylation (not shown).[8]
As part of their investigations,Amrein and Studer
demonstrated the possibility of radical transfer hydrosilyla-
tion of aldehydes (Scheme 5).[8] These reactions had to be
performed at 14088Cinasealed tube with di-tert-butyl
peroxide as initiator. Alkyl aldehydes 15 and even cyclo-
hexanone (not shown) reacted readily under these conditions
(15!16). Conversely,aryl aldehydes 17,for example,
benzaldehyde,were not converted into 18,presumably
because of the stability of the intermediate benzyl radical,
which cannot be reduced by cyclohexa-1,4-diene 1.
3. Ionic Transfer Hydrosilylation
Theorigin of ionic transfer hydrosilylation can be seen in
the B(C6F5)3-promoted hydride abstraction from 1,4-dihydro-
pyridines (Scheme 6). Stephan, Crudden, and co-workers
systematically investigated the abstraction of hydride from
Hantzsch-type dihydropyridines by the strong Lewis acid
B(C6F5)3.[9] Clean formation of the pyridinium ion with
[HB(C6F5)3]¢as counteranion was found for Hantzsch
hydride donors with an NMe rather than afree NH group
(19!20,Scheme 6, top). An experiment hidden in publica-
tions by Nikonov and co-workers is even more pertinent to
ionic transfer hydrosilylation.[10] Hydride abstraction from N-
silylated 1,4-dihydropyridine 21 generated the ion pair 22
(Scheme 6, bottom). Adduct 22 is a(reversible) frustrated
Lewis pair/hydrosilane system[11] composed of pyridine/
B(C6F5)3and Me2PhSiH, from which the hydrosilane is slowly
released (22!23,Scheme 6, bottom).
NikonovÏs experiment is intriguing in the sense that the
electron-deficient borane employed for the hydrosilane
release from the partially reduced silicon-substituted hetar-
ene is also asuperb Lewis acid for the activation of Si¢H
bonds.[12] Abroad spectrum of hydrosilylation reactions is in
fact catalyzed by B(C6F5)3.[13] However,pyridine and B(C6F5)3
form Lewis pair 23,which will hamper the subsequent Si¢H
bond activation. This obstacle would be overcome by using 3-
silylated cyclohexa-1,4-dienes instead of N-silylated 1,4-
dihydropyridines.The arene formed as waste in the hydro-
silane release step would not interfere with B(C6F5)3-cata-
lyzed hydrosilylations.The overall strategy,therefore,consists
of two consecutive catalytic cycles,both promoted by B-
(C6F5)3(Scheme 7).[14] Aquantum-chemical treatment by
Sakata and Fujimoto later confirmed the mechanism pro-
posed for p-basic alkenes by Simonneau and Oestreich.[15] It
still needs to be verified whether the concecutive process also
applies to s-basic aldehydes as these could competitively
Scheme 4. Radical transfer hydrosilylation coupled with radical cycliza-
tion.
Scheme 6. Hydride abstraction from 1,4-dihydropyridines by B(C6F5)3.
Scheme 7. Consecutive catalyticcycles of ionic transfer hydrosilylation.
X=CH2(alkenes) and O(aldehydes).
Scheme 5. Radical transfer hydrosilylation of aldehydes.
A
ngewandte
Chemie
Minireviews
496 www.angewandte.org Ó2016 The Authors. Published by Wiley-VCH Verlag GmbH &Co. KGaA, Weinheim Angew.Chem. Int. Ed. 2016,55,494 –499
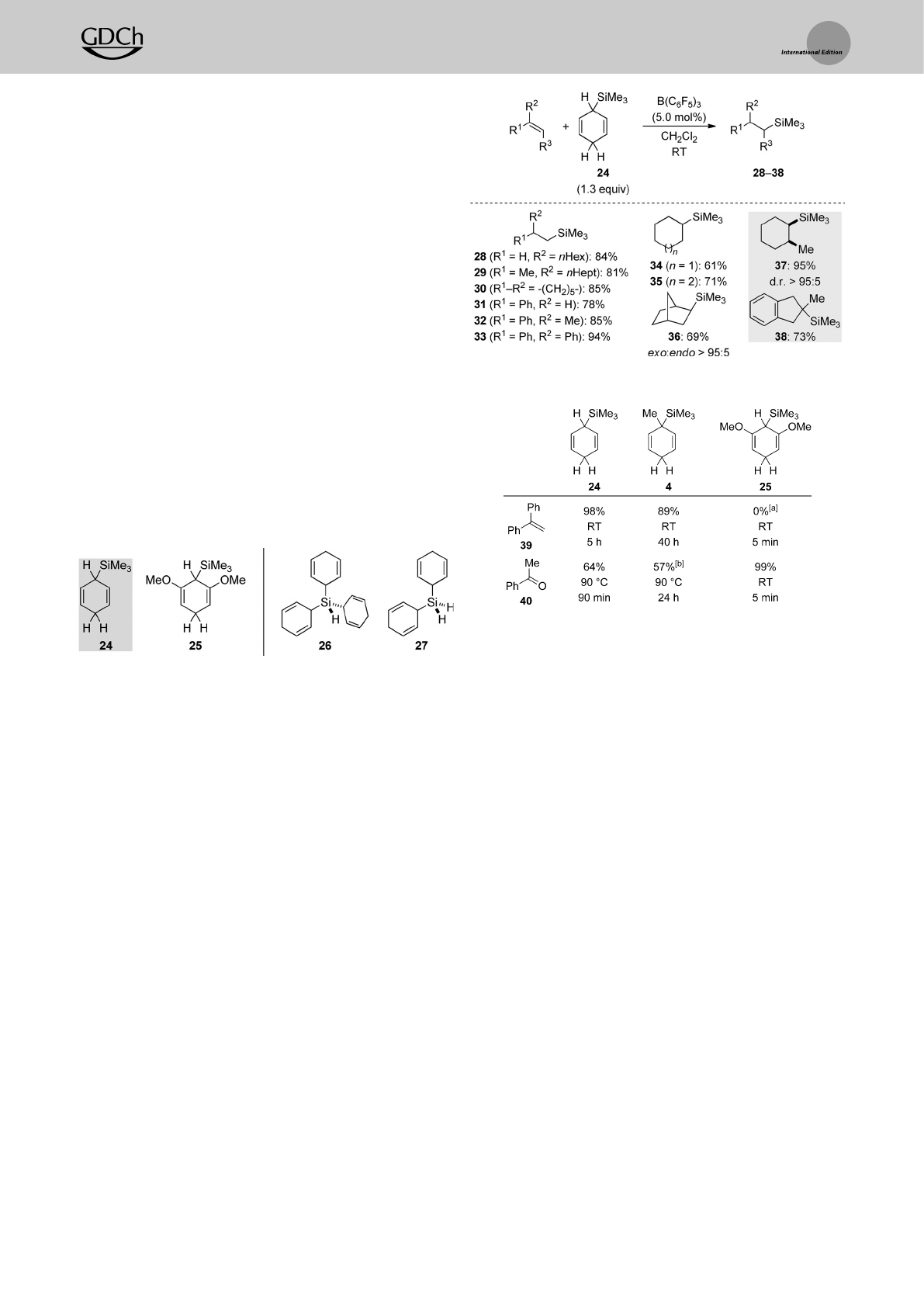
capture the silicon electrophile at an earlier stage that is
interrupting the hydrosilane-release cycle.
Thehydrosilane-release cycle (Scheme 7, left) commen-
ces with coordination of B(C6F5)3to the methylene C¢H
group opposite to the face with the silicon group (XIII!
XIV).[15] This reversible interaction eventually leads to
hydride abstraction and formation of silicon-stabilized cyclo-
hexadienyl cation XV (XIV!XV).[3] Intermediate XV was
our source of inspiration, as we had initially been interested in
making arene-stabilized silicon cations such as XV by hydride
abstraction from cyclohexa-2,5-dien-1-yl-substituted silanes
XIII with the trityl cation (the “cyclohexadienyl-leaving-
group” approach).[16] Neutral B(C6F5)3yields,however,[HB-
(C6F5)3]¢,which reduces the silicon cation in ion pair XV to
afford the hydrosilane and arene XVI.That hydrosilane then
enters the hydrosilylation cycle (Scheme 7, right) to form
adduct XVII[12] in equilibrium. TheB(C6F5)3-activated hydro-
silane XVII reacts with various p-and s-basic substrates X,
for example,alkenes[17] and aldehydes[18].Formal transfer of
the silicon cation onto the Lewis base is followed by
borohydride reduction (X!XVIII!XII).
To test this above strategy,several cyclohexa-1,4-diene-
based hydrosilane surrogates were prepared, for example, 24
and 25 (Figure 2, left).[14,19] Variation of the substitution
pattern at the silicon atom was also investigated, but the focus
of Oestreich and co-workers was on the transfer of otherwise
gaseous hydrosilanes such as Me3SiH, Me2SiH2,and difficult-
to-handle SiH4.26 and 27 are such surrogates for monosilane
(Figure 2, right).[20]
Theionic transfer hydrosilylation emerged as broadly
applicable to unfunctionalized alkenes (Scheme 8).[14] The
reaction conditions were mild, just maintaining the reactants
together with the catalyst in CH2Cl2[15] at room temperature.
Terminal, that is,mono- and 1,1-disubstituted, alkenes reacted
smoothly (!28–33), as did internal alkenes (!34–38). The
exo selectivity seen for norbornene (!36)and the predom-
inant cis diastereoselectivity in the hydrosilylation of 1-
methylcyclohexene (!37)were evidence of the involvement
of carbenium ion intermediates.The hydrosilylation of
another trisubstituted alkene,2-methyl-1H-indene,show-
cased the better stabilization of abenzylic compared to
atertiary carbenium ion (!38).
Oestreich and co-workers recently reported asystematic
study of the ionic transfer hydrosilylation.[19,21] This work
includes screenings of representative p-and s-donating
substrates,electronically and sterically modified surrogates,
and partially or fully fluorinated triarylboranes.Selected
representative data are depicted in Scheme 9. Thereduction
of s-basic acetophenone (40)using standard 24 requires
higher temperature than that of p-basic 1,1-diphenylethylene
(39)because 40 forms astronger Lewis acid/base adduct with
B(C6F5)3(column 1). Cognate 4,previously used by Studer
and co-workers in the radical transfer hydrosilylation,[7] with
an additional methyl group at the silicon-bearing carbon atom
is less reactive (column 2). Resorcinol-derived 25,which
mimics StuderÏs reagents (cf.Figure 1), is far more reactive
than 24 because of its enhanced hydricity (column 3). How-
ever,the Lewis-basic methoxy groups in 25 compete with the
substrate for the transfer of the silicon electrophile (cf.
XVII!XVIII,Scheme 7). The p-basic alkene is not suffi-
ciently nucleophilic,and demethylation of the resorcinol
dimethyl ether was observed. Conversely,the carbonyl
hydrosilylation was complete at room temperature within
minutes.
Acrucial test of transfer hydrosilylation is whether it
would enable the transfer of monosilane.The serious safety
issues associated with handling SiH4have deterred synthetic
chemists from its use,and conventional hydrosilylation with
this dangerous gas is barely researched. Simonneau and
Oestreich introduced solid 26 and liquid 27 as monosilane
surrogates to ionic transfer hydrosilylation (Figure 2, right
and Scheme 10).[20] It was shown that B(C6F5)3unleashes SiH4
Figure 2. Silicon-substituted cyclohexa-1,4-dienes for ionic transfer
hydrosilylation.
Scheme 8. Ionic transfer hydrosilylation of alkenes.
Scheme 9. Gauging the reactivity of surrogate/substrate combinations
in ionic transfer hydrosilylation. [a] 25 completely consumed. [b] Partial
deoxygenation to styrene.
A
ngewandte
Chemie
Minireviews
497Angew.Chem. Int.Ed. 2016,55,494 –499 Ó2016 The Authors. Published by Wiley-VCH Verlag GmbH &Co. KGaA, Weinheim www.angewandte.org
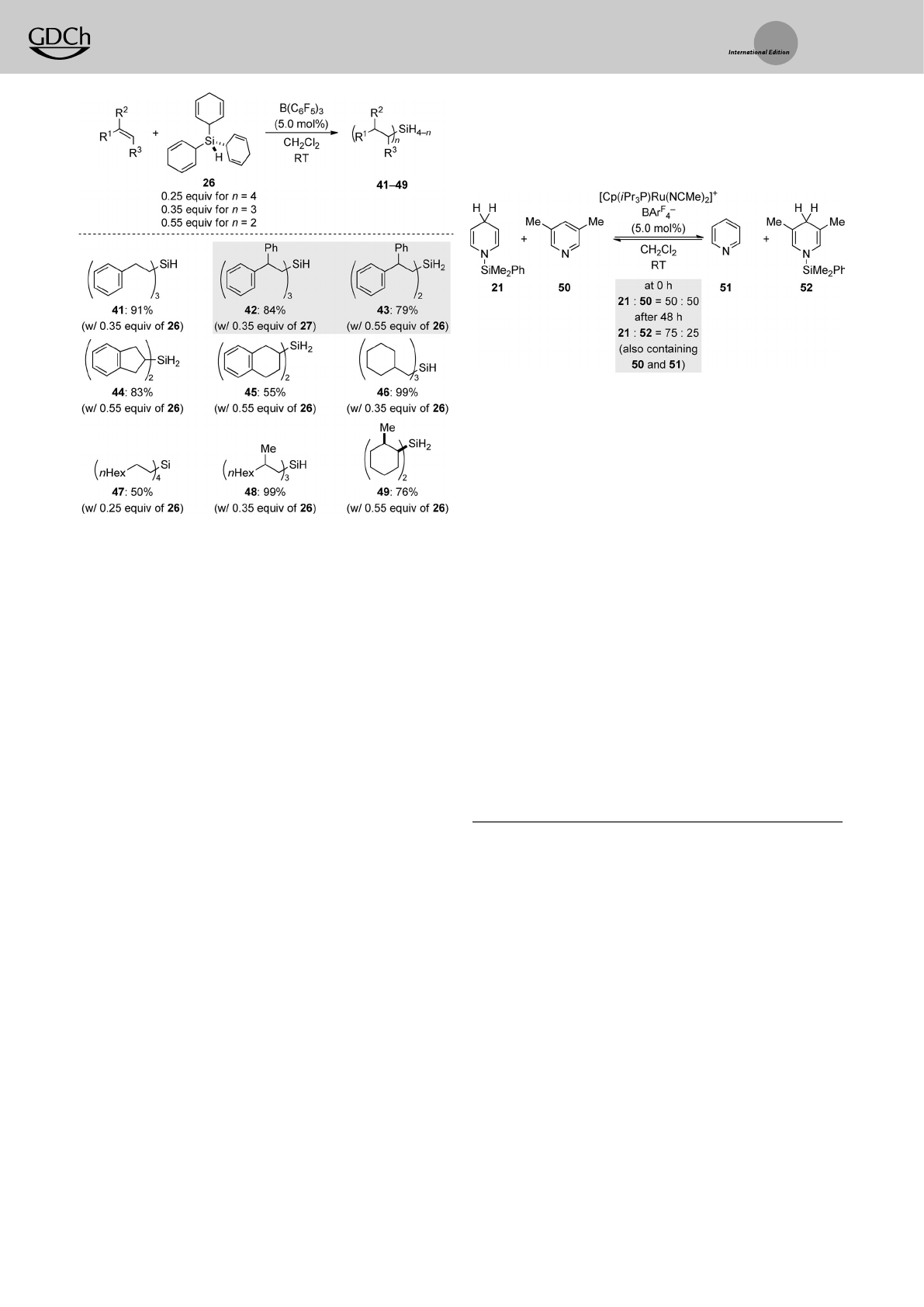
prior to n-fold hydrosilylation of typical alkenes.The chemo-
selectivity is determined by steric demand and cannot be
controlled by the ratio of the reactants.Inone case,the use of
27 (0.35 equiv) or 26 (0.55 equiv) led to the selective
formation of monohydrosilane 42 (n=3) and dihydrosilane
43 (n=2), respectively (gray box). An a-olefin afforded the
tetraorganosilane (!47), but the other transfer reactions
yielded either mono- or dihydrosilanes with synthetically
useful selectivities,including styrene (!41).
4. Outlook
Transfer hydrosilylation is at the early stages of develop-
ment, but the variants disclosed by the groups of Studer[7,8]
and Oestreich[14,19,20] have already indicated its potential. The
radical process is superior to B(C6F5)3catalysis because of its
compatibility with Lewis-basic functional groups,carboxyl
groups in particular.Moreover,the ionic pathway cannot
transfer silicon groups with bulky substituents,for example,
tBuMe2Si and iPr3Si.[14] Thereaction temperature of the
cationic process is,however, an advantage when transferring
gaseous and hence difficult-to-handle hydrosilanes,for exam-
ple,Me
3SiH, Me2SiH2,and SiH4.The focus of the work of
Simonneau and Oestreich is on these pyrophoric and
explosive hydrosilanes,and the monosilane surrogates finally
allow safe synthetic chemistry with this hazardous smallest
member of the hydrosilane family.
These transfer hydrosilylations are transition-metal free,
and this naturally raises the question whether transition
metals are also able to catalyze the transfer of ahydrosilane
from one molecule to another.Reversibility of the hydro-
silylation would be abasic requirement for this.Itwas again
the groups of Nikonov[10] and later Oestreich[22] who demon-
strated such reversibility for 1,4-selective pyridine hydro-
silylation (Scheme 11). NikonovÏs fundamental discovery is
again buried in his method-oriented publications (cf.
Scheme 6, bottom). Aruthenium catalyst promotes hydro-
silane transfer from an N-silylated 1,4-dihydropyridine to 3,5-
lutidine (21!52,Scheme 11);anitrile also served as an
acceptor (not shown).[10] This proof of concept of atransition-
metal-catalyzed transfer hydrosilylation could prove viable in
the future.
Acknowledgements
This research was supported by the Deutsche Forschungsge-
meinschaft (Oe 249/11-1). M.O.isindebted to the Einstein
Foundation (Berlin) for an endowed professorship.Ithank
Dr. Antoine Simonneau and Sebastian Keeß for their
enthusiasm and commitment.
Howtocite: Angew.Chem. Int. Ed. 2016,55,494–499
Angew.Chem. 2016,128,504–509
[1] D. Wang,D.Astruc, Chem. Rev. 2015,115,6621 –6686.
[2] A. Studer,S.Amrein, Angew.Chem. Int. Ed. 2000,39,3080–
3082; Angew.Chem. 2000,112,3196–3198.
[3] These silicon-substituted cyclohexadienyl cations are low-energy
Wheland complexes or, from the perspectiveofsilicon chemis-
try,arene-stabilized silicon cations:J.B.Lambert,S.Zhang,
C. L. Stern, J. C. Huffman, Science 1993,260,1917–1918.
[4] M. Kira, H. Sugiyama, H. Sakurai, J. Am. Chem. Soc. 1983,105,
6436–6442.
[5] A. Studer,S.Amrein, F. Schleth, T. Schulte,J.C.Walton, J. Am.
Chem. Soc. 2003,125,5726 –5733.
[6] J. C. Walton, A. Studer, Acc. Chem. Res. 2005,38,794 –802.
[7] a) S. Amrein, A. Timmermann, A. Studer, Org.Lett. 2001,3,
2357–2360;b)S.Amrein, A. Studer, Chem. Commun. 2002,
1592–1593.
[8] S. Amrein, A. Studer, Helv.Chim. Acta 2002,85,3559–3574.
[9] J. D. Webb,V.S.Laberge,S.J.Geier,D.W.Stephan, C. M.
Crudden, Chem. Eur.J.2010,16,4895–4902.
[10] a) D. V. Gutsulyak, A. van der Est, G. I. Nikonov, Angew.Chem.
Int. Ed. 2011,50,1384–1387; Angew.Chem. 2011,123,1420 –
Scheme 10. Ionic transfer hydrosilylation of alkenes with monosilane.
Scheme 11. Reversiblehydrosilylation of pyridines: An example of
atransition-metal-catalyzed transfer hydrosilylation. BArF4¢=tetrakis-
[3,5-bis(trifluoromethyl)phenyl]borate.
A
ngewandte
Chemie
Minireviews
498 www.angewandte.org Ó2016 The Authors. Published by Wiley-VCH Verlag GmbH &Co. KGaA, Weinheim Angew.Chem. Int. Ed. 2016,55,494 –499
Loading more pages...