
Mater. Res. Express 9(2022)065101 https://doi.org/10.1088/2053-1591/ac784f
PAPER
Ion-mediated desorption of asphaltene molecules from carbonate
and sandstone structures
Pouyan Ahmadi
1
, Mohammadreza Aghajanzadeh
2,3
, Hamidreza Asaadian
4,∗
, Armin Khadivi
5
and
Shahin Kord
6
1
Department of Hydrogeology, Helmholtz-Centre for Environmental Research, UFZ, Germany
2
Department of Civil Engineering, Monash University, Melbourne, Australia
3
Department of Petroleum Engineering, Amirkabir University of Technology, Tehran, Iran
4
Norwegian University of Science and Technology (NTNU), Department of Geoscience and Petroleum, Trondheim, Norway
5
Technical University of Berlin, Berlin, Germany
6
Ahwaz Faculty of Petroleum, Petroleum University of Technology, Ahwaz, Iran
∗
Author to whom any correspondence should be addressed.
Keywords: Wettability state, Molecular dynamic simulation, Asphaltene precipitation, Smart water, Bonding and non-bonding energy,
Coulomb interaction
Abstract
As more and more oil recovery scenarios use seawater, the need to identify the possible mechanisms of
wettability state changes in oil reservoirs has never been greater. By using molecular dynamics
simulations, this study sheds light on the effect of ions common to seawater (Ca
2+
,K
+
,Mg
2+
,Na
+
,
Cl
−
, HCO
3−
,SO
4
2−
)on the affinity between silica and carbonate as the traditional rock types and
asphaltene molecules as an important contributing factor of reservoir oil wetness. In the case of
carbonate and silica being the reservoir rock types, the measured parameters indicate good agreement
with each other, meaning that (HCO
3
−
&SO
4
2−
)and(Na
+
&Cl
−
)ions reached maximum bonding
energies of (25485, 25511, 4096, and −4093 eV, respectively). As with the surface charge density
measurements, the results of the non-bonding energies between the individual atomic structures agree
with those from the simulation cell. In the presence ofa silica surface, the radial distribution function
(RDF)results determine that the peak of the maximum value for the distribution of theions is 4.2.
However, these values range from 3 to 6.6, suggesting that different ions perform better under the
influence of carbonate rock. As these ions are distributed in the simulation box along with the
adsorption domain, the conditions for sequestering asphaltene from the rock surface are made ideal
for dissolution and removal. At equal ion strength, measuring the distance between the center of mass
of rocks and asphaltene structures reveals a maximum repulsion force of 22.1 Å and a maximum
detachment force of 10.4 Å in the presence of SO
4
2−
and Na
+
ions on carbonate and silica surfaces.
1. Introduction
Smart water flooding has been proven to be one of the most successful methods for improving oil recovery due
to the impact of water chemistry and salinity on reservoir fluid-fluid and rock-fluid interactions [1–5]. It has
been shown that the recovery of crude oil can be improved by manipulating the injection water [6–9]. Although
numerous studies have been conducted over the last two decades to study the effect of smart salinity water
flooding on the improvement of crude oil recovery in carbonate and sandstone reservoirs, the underlying
mechanisms have not been completely revealed [2]. Multi-ion exchange (MIE), double-layer expansion (DLE),
wettability alteration, and pH effect are the most important mechanisms that have been widely reported in the
literature. Among these mechanisms, wettability alteration from oil-wet to water-wet is a widely accepted
mechanism in this flooding method [10–17].
The wettability of the pore surface plays a vital role in the displacement of the crude oil by water. The
wettability state of the reservoir rock surface can be attributed to the adsorption of polar components from crude
OPEN ACCESS
RECEIVED
6 April 2022
REVISED
9 June 2022
ACCEPTED FOR PUBLICATION
13 June 2022
PUBLISHED
28 June 2022
Original content from this
work may be used under
the terms of the Creative
Commons Attribution 4.0
licence.
Any further distribution of
this work must maintain
attribution to the
author(s)and the title of
the work, journal citation
and DOI.
© 2022 The Author(s). Published by IOP Publishing Ltd

oil at the mineral surfaces [18–22]. It is generally accepted that underground reservoirs are saturated with
formation brine before oil migrates into their rock pores. After the drainage of water by crude oil from the
porous media, the reservoir wettability may become less water-wet or even oil-wet because of the adsorption of
heavy components of crude oil onto rock surfaces. The main mechanisms by which crude oil compounds may
adsorb onto pore surfaces include surface precipitation, polar interactions, and acid/base and ion binding
interactions [18,23,24].
Asphaltenes are the most polar fraction in crude oil [25,26]. The adsorption of asphaltenes on mineral
surfaces is an important element of wettability changes toward oil-wet conditions and can have a strong negative
impact on rock properties. Asphaltenes are polycyclic aromatic hydrocarbons (PAHs)consisting of a sheet-like
structure of interlocked heterocyclic aromatic rings attached to hydrocarbon chains andcontaining both polar
and non-polar species [27]. In addition, heteroatoms such as oxygen, sulfur, and nitrogen atoms and trace
amounts of metals such as Fe, Ni, and V in asphaltene molecules make these molecules the most polar and
complex components of crude oil [28]. Changing the thermodynamic conditions may cause the precipitation
and adsorption of asphaltene onto the rock surface during the production and transportation of crude oil. The
adsorption of precipitated asphaltenes onto the rock surface can lead to formation damage in oil reservoirs by
reducing the effective oil permeability [29–31].
Many researchers have investigated the adsorption/desorption process of asphaltene molecules on and from
rock surface [31–36]. It was found that precipitated asphaltene molecules on the rock surface can be adsorbed in
nanofluids containing metal oxide nanoparticles, such as TiO
2
, SiO
2
and Fe
2
O
3
[30,37,38]. Other experimental
and modeling studies have revealed that the injection of modified brine during the EOR process could desorb
asphaltene molecules and other polar components of crude oil from the mineral surfaces and alter the
wettability of the pore wall to less oil wet, resulting in improved oil recovery [7,39–42]. Ligthelm et al. showed
that a decrease in salinity increased the expansion of the diffuse double layer between the rock and oil interfaces,
facilitating the release of organic materials [11]. Yang et al. studied the desorption of asphaltenes from quartz
crystals in the presence of an electrolyte. They used a quartz crystal microbalance with dissipation (QCM-D),
atomic force microscopy (AFM), and contact angle to measure the amount of desorbed asphaltenes. They found
that desorption occurred when the rock surface was exposed to a saline solution. This is mainly because the
charge density of both the surfaces of oil-water and solid-water was promoted and the electrostatic repulsions
increased [43].
Despite extensive experimental and modeling studies on the effect of salinity on the adsorption/desorption
of asphaltene molecules from mineral surfaces, the exact mechanism that occurs at the atomic scale still needs to
be investigated in more detail; hence, the present work has concentrated on checking the influence of ions on the
absorbance tendency of polar asphaltene molecules on the surface of sandstone and carbonate rocks under
reservoir conditions. Hence, molecular dynamics simulations were used to understand the behavior of
asphaltene molecules upon exposure to cations and anions (i.e., Ca
2+
,K
+
,Mg
2+
,Na
+
,Cl
−
, HCO
3
−
,SO
4
2−
)
available in smart water (salinity5000 ppm)in the presence of carbonate and sandstone rock types [44]. The
LAMMPS (stable release)software was used as the simulation tool, and all numerical calculations were crunched
using this computational package. All investigation methods, such as surfacecharge density measurement,
radial distribution function (RDF), discretizing of bonding and non-bonding energies, and measuring the
asphaltene detachment distance, have proved the importance of ion types on the fate of asphaltene molecules
that are stuck to the rock surface in reservoirs.
2. Computational method
2.1. Force field and relative parameters
The present work employs molecular dynamics (MD)simulations to investigate the precipitation of asphaltene
on carbonate and sandstone structures in the presence of brine at a defined temperature and pressure. MD
simulation is known as a computational method to estimate the dynamic translocation of particles considering
the underlying concept of intermolecular interactions at the atomic scale. In addition to the molecular
interaction for a specific time interval (time step), this method can provide an exact insight into the particle
transformation of the structures. In this method, Newton’s law, which is a numerically solving method,
determines the particle trajectories. Likewise, the interaction between the particles and their interatomic
energies is often computed using force fields in the system. In this study, Large-scale Atomic/Molecular
Massively Parallel Simulator (LAMMPS)software was used as a simulation tool, and the entire calculation of
asphaltene deposition on carbonate and sandstone structures was implemented by this computational package
using the Lennard Jones (LJ)potential [45]. This software was developed and released by Sandia National
Laboratories (SNL). Simulations of this research were carried out in the following steps, as mentioned
previously:
2
Mater. Res. Express 9(2022)065101 P Ahmadi et al

Step 1: In this step, the preliminaries of the simulation are set up, such as units, boundary conditions,
dimensions, bond type, atom location, and structure style. In the following simulations, the units were adjusted
real, the boundary condition was periodic, and the atomic structure was full.
Step 2: In the next step, atoms and molecules were introduced into the simulation environment. After
packing molecules with another software named Packmole and obtaining their data files, they were presented as
input to the designated simulation cell by the read data command. Furthermore, specific groups of atoms (i.e.,
Ca
2+
,K
+
,Mg
2+
,Na
+
,Cl
−
, HCO
3
−
,SO
4
2−
)were assigned and distinguished with unique names by group
commands to achieve better control over them during simulations. In this stage, everything is well defined and
the software is ready to start the simulation.
Step 3: In the last step, an approximate velocity must be estimated to minimize the convergence time by the
velocity command before reaching system stability. The more related system velocity according to the system
convergence temperature was estimated so that the computational time of the software used reached its
minimum as much as possible. The convergence temperature was set to 60°C or 333.15 K for all scenarios to
resemble reservoir conditions.
Pairstyle commands outline interactions (potential of forces)between atoms and molecules, such as
intermolecular, bond, and nonbonding energies. All interaction calculations were based on the Lennard-Jones
equation in the following simulation work.
Lennard-Jones presented the first formula for this relationship in 1924 [45]. The following equation is called
the Lennard (LJ)potential.
⎜⎟ ⎜⎟
⎡
⎣
⎢⎛
⎝
⎞
⎠
⎛
⎝
⎞
⎠
⎤
⎦
⎥
=e s-s<rrr
rrU4 1
ij ij
c
12 6
() ()
where U(r)represents the Dreiding force field [45], is the non-bond interaction, εis the depth of the potential
well, σis the distance at which the potential is 0, and r
ij
is the distance between the atoms and molecules. This
equation can theoretically estimate the interactions between a pair of particles (atoms and molecules). Table 1
presents the scale of the length and energy parameters of various atoms in the simulated structures. The cutoff
radius was set to 12 Å for interactions between pairs of particles. Furthermore, Coulomb interaction was
implemented for the simulated structures using the following equation:
e
=<rrrrECQ Q 2
c
ij
() ()
Where C is the energy-conversion constant, and εrepresents the dielectric constant. Q
i
and Q
j
are the charges on
two atoms. The cut-off radius is set to 10 Å forthis type of non-bonding interaction, and all atomic structures
with distances larger than the cut-off radius will not be taken into account in the numerical calculations
<rr.
c
(
)
The bond and angle strengths for the Dreiding potential are defined by a simple harmonic oscillator
equation as follows:
=-
q
EKrr12 3
r2
() ()
=-EKrr12 4
r02
() ()
where K
r
and K
θ
are harmonic oscillator constants, and r
0
and θ
0
represent the length of the atomic bondand
equilibrium value of the angle, respectively. In this work, the harmonic oscillator constants were adjusted to 700
((kcal/mol)/Å
2
)and 100 ((kcal/mol)/degree
2
)for K
r
and K
θ
, respectively. The atomic bond lengths and
Table 1. The εand σparameters for
non-bond interactions in simulated
atomic structure [45].
ε(kcal/mol)σ(Å)
C 0.1450 3.9800
H 0.0100 3.20000
O 0.4150 3.71
S 0.3050 4.24
N 0.4150 3.9950
Ca 0.0500 3.4720
Si 0.0950 4.4350
Cl 0.3050 3.9150
F 0.3050 3.2850
K 0.3050 3.398
Mg 0.111 2.692
Na 0.5 3.144
3
Mater. Res. Express 9(2022)065101 P Ahmadi et al
equilibrium values of the angles for each bonded interaction are listed in table 2[45]. Furthermore, the dihedral
interactions in these simulations were based on previously reported bonding interactions. These bonding
interactions were calculated using the harmonic equation, and the coefficients were selected from the Dreiding
force field [45]:
=+ ÆEK dcosn15(()) ()
where K is the harmonic oscillator constant, d=+1or−1 and n is the integral number [45].
In addition, the Dreiding force field is used for rock-asphaltene, rock–ion, ion–asphaltene, and rock-fluid
interactions. Moreover, Newton’s second law at the atomic level was used for computations of the particle
motion through the simulation time. The following formula shows the gradient of the potential relation:
å
== = =-
¹
Frv
Fm
d
dt md
dt grad V r 7
ij i
i
ij
iiiij
2
2
() ()
The association of previous relations is performed using the velocity-Verlet algorithm to integrate Newton’s law
as follows:
dd+= +vttvtatt 8( ) () () ()
dd+= +vttrtvtt 9( ) () () ()
In these two relations, r(t+δt)and v(t+δt)are the final position and velocity of the atoms (respectively)and r
(t)and v(t)are the initial rates of these mechanical parameters.
2.2. Designed atomic structures of molecules in the simulation cell
The designed simulation cells containing carbonate and sandstone surfaces along with the ions and asphaltene
molecules were adjusted according to the Dreiding force field parameters to define the atomic structures and
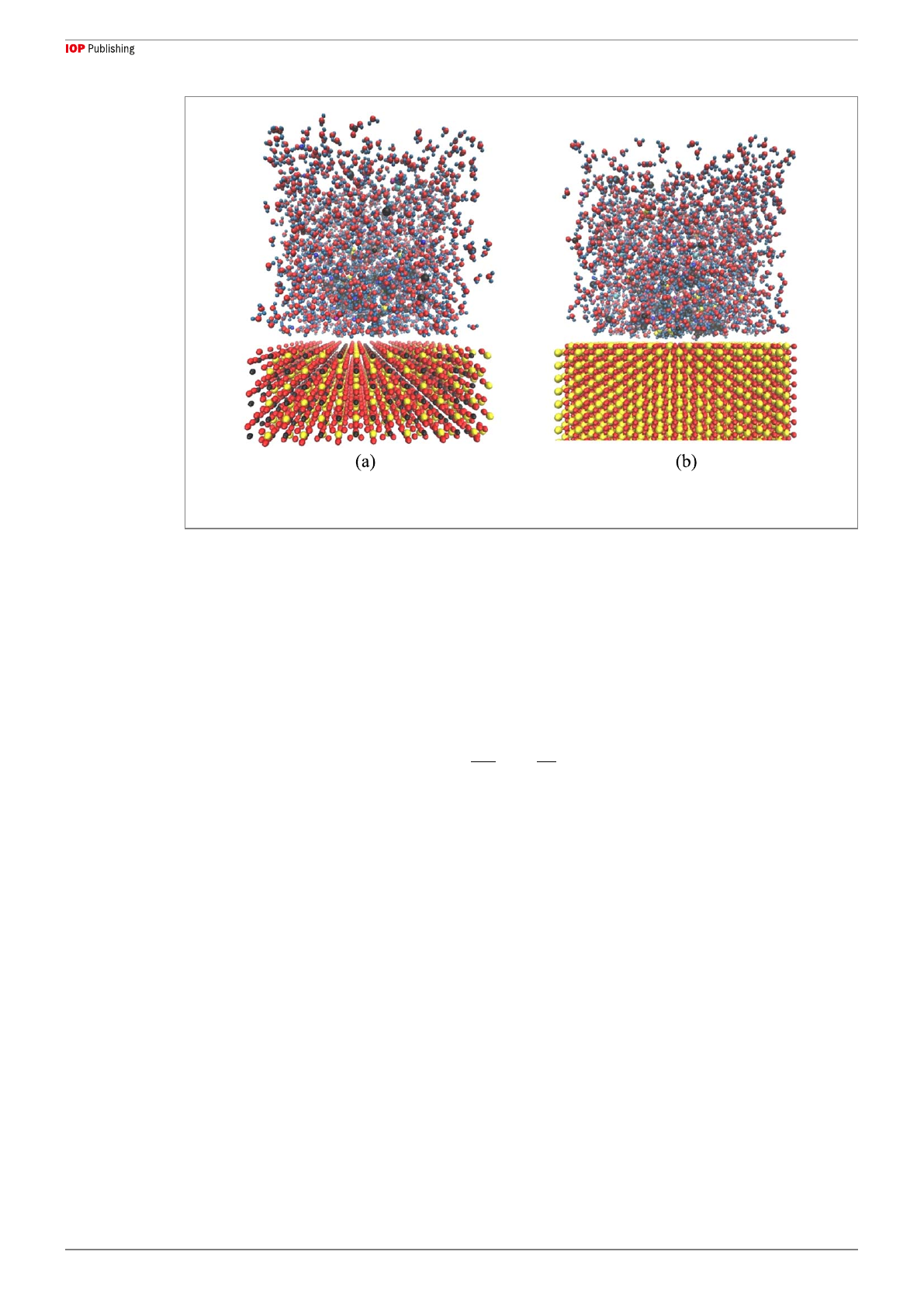
force field. Figure 1shows the frame in which the studied ions were randomly arranged by Packmole software
(version 20.010)in such a way that the ion strength for all scenarios was kept constant with a view to having an
exact comparison of ion impact on the way rock that the asphaltene molecule deals with rock surfaces in all cases.
In support of this idea, 10 asphaltene molecules considered in all scenarios and 100 of each ion were randomly
distributed in the designed cell, and the effect of the mixture of all ions was examined to observe the behavior of
asphaltene. Moreover, because most of the carbonate and sandstone rock constituents are made of SiO
2
and
CaCO
3
, respectively, and to eliminate the impact of impurities, the pure structures of each rock type were
considered in all simulations.
In all scenarios, water molecules were also added to thecells considering their real number, which was
proportional to the designed simulation cell volume. The pressure and temperature were adjusted to 60°C and
2500 psi, respectively, to make the situation more thermodynamically similar to real reservoir conditions. In this
study, the asphaltene molecule is precipitated due to thermodynamic disturbance and is not soluble in the water
Figure 1. Atomic structures with minimum tolerance of 2 Angstrom, a)Oil wet carbonate surface surrounded by common active ions
of seawater (left).b)Oil wet sandstone surface surrounded by common active ions of seawater (right).
4
Mater. Res. Express 9(2022)065101 P Ahmadi et al

bulk, which implies that the asphaltene molecule detachment could either intensify or alleviate under the
influence of what happens in the brine solution. Figure 2shows the structure of the polar asphaltene molecules,
which was used to check the effect of their tendency to be adsorbed on the rock surfaces. The asphaltene
structure belongs to one of the southern Iranian oilfields, which contains heteroatoms (nitrogen, oxygen, and
sulfur)as the electronegative part of the asphaltene molecule.
3. Results and discussion
This study investigated a wide variety of possible aspects that could explain the interaction tendency between
rock, asphaltene molecules, and ions. The appropriate parameters that could reveal the affinity between
individual structures in an atomic-scale simulation cell, including bonding and non-bonding energies, surface
charge density, asphaltene angle on the rock, ion density inside the simulation cell, radial distribution function
(RDF), and displacement of the atomic structure center, will be discussed in order to gain a thorough
understanding of the behavior of asphaltene molecules on carbonate and sandstone surfaces. These surfaces
have been selected because they comprise the majority of conventional and unconventional rock types
worldwide.
Table 2. The equilibration distance/
angle for bond strength and bond-angle
bend in MD simulations [45].
Parameter r
0
(Å)θ
0
(degree)
C-C bond 1.530 —
N-F bond 1.371 —
C-H bond 1.090 —
C-N bond 1.462 —
C-O bond 1.420 —
C-S bond 1.800 —
C-Cl bond 1.757 —
H-N bond 1.022 —
H-O bond 0.980 —
H-S bond 1.360 —
O-S bond 1.690 —
O-Si bond 1.587 —
S-S bond 2.070 —
C-C-C —109.471
C-C-F —109.471
C-C-H —109.471
C-C-N —109.471
C-C-O —109.471
C-H-S —180.000
C-N-C —106.700
C-N-H —106.700
C-S-C —92.100
C-S-H —92.100
C-S-O —92.100
C-S-S —92.100
H-C-H —109.471
H-C-N —109.471
H-C-S —109.471
H-O-H —104.51
H-S-H —92.100
H-S-O —92.100
H-S-S —92.100
N-C-O —109.471
O-S-S —92.100
O-Si-O —109.471
S-C-S —109.471
S-O-S —104.51
Si-O-Si —104.51
5
Mater. Res. Express 9(2022)065101 P Ahmadi et al
Loading more pages...