
Citation: Tsukanov, A.A.; Shilko, E.V.;
Popov, M. Structure, Properties, and
Phase Transformations of Water
Nanoconfined between Brucite-like
Layers: The Role of Wall Surface
Polarity. Materials 2022,15, 3043.
https://doi.org/10.3390/
ma15093043
Academic Editor:
Edward Bormashenko
Received: 30 March 2022
Accepted: 20 April 2022
Published: 22 April 2022
Publisher’s Note: MDPI stays neutral
with regard to jurisdictional claims in
published maps and institutional affil-
iations.
Copyright: © 2022 by the authors.
Licensee MDPI, Basel, Switzerland.
This article is an open access article
distributed under the terms and
conditions of the Creative Commons
Attribution (CC BY) license (https://
creativecommons.org/licenses/by/
4.0/).
materials
Article
Structure, Properties, and Phase Transformations of Water
Nanoconfined between Brucite-like Layers: The Role of Wall
Surface Polarity
Alexey A. Tsukanov 1,* , Evgeny V. Shilko 2,* and Mikhail Popov 3
1
Center for Computational and Data-Intensive Science and Engineering (CDISE), Skolkovo Institute of Science
and Technology (Skoltech), 121205 Moscow, Russia
2Institute of Strength Physics and Materials Science of SB RAS, 634055 Tomsk, Russia
3Institut für Mechanik, Technische Universität Berlin, 10623 Berlin, Germany; [email protected]
*Correspondence: [email protected] (A.A.T.); [email protected] (E.V.S.)
Abstract:
The interaction of water with confining surfaces is primarily governed by the wetting
properties of the wall material—in particular, whether it is hydrophobic or hydrophilic. The hy-
drophobicity or hydrophilicity itself is determined primarily by the atomic structure and polarity
of the surface groups. In the present work, we used molecular dynamics to study the structure
and properties of nanoscale water layers confined between layered metal hydroxide surfaces with
a brucite-like structure. The influence of the surface polarity of the confining material on the prop-
erties of nanoconfined water was studied in the pressure range of 0.1–10 GPa. This pressure range
is relevant for many geodynamic phenomena, hydrocarbon recovery, contact spots of tribological
systems, and heterogeneous materials under extreme mechanical loading. Two phase transitions
were identified in water confined within 2 nm wide slit-shaped nanopores: (1) at p
1
= 3.3–3.4 GPa, the
liquid transforms to a solid phase with a hexagonal close-packed (HCP) crystal structure, and (2) at
p
2
= 6.7–7.1 GPa, a further transformation to face-centered cubic (FCC) crystals occurs. It was found
that the behavior of the confined water radically changes when the partial charges (and, therefore,
the surface polarity) are reduced. In this case, water transforms directly from the liquid phase to
an FCC-like phase at 3.2–3.3 GPa. Numerical simulations enabled determination of the amount
of hydrogen bonding and diffusivity of nanoconfined water, as well as the relationship between
pressure and volumetric strain.
Keywords:
nanoconfined water; nanopore; interface; surface polarity; phase transformation; layered
metal hydroxide; high-pressure crystallization; molecular dynamics
1. Introduction
Solid and liquid phases confined to a volume of several atomic sizes (nanoconfined
matter) may significantly or even qualitatively differ from the corresponding bulk phases.
This is valid both for nanosized particles or droplets and for liquids or solids confined
within nanosized pores or gaps [
1
–
5
]. Under conditions of nanoconfinement, many kinetic
and thermodynamic processes—including chemical reactions—demonstrate extraordinary
behavior not observed in bulk conditions [
6
–
8
]. This causes both nanoconfined matter and
processes occurring under low-dimensional constraints to be of increasing interest in a
variety of scientific and industrial areas [
1
,
9
,
10
]. For example, nanoconfined hydrides are
among the most promising solutions for hydrogen storage [
11
]. Confined ionic liquids
are used in promising supercapacitors (electrochemical devices that store energy through
the reversible adsorption of ionic species) with enhanced capacitance and the capability
of fast charging and discharging [
12
,
13
]. Nanoconfinement-based methods are used for
the analysis of long biomolecules [
14
,
15
], as well as for studies of hydrocarbon-recovery-
related problems using nanofluidic devices [
16
,
17
]. Matter in nanoconfined conditions is
Materials 2022,15, 3043. https://doi.org/10.3390/ma15093043 https://www.mdpi.com/journal/materials

Materials 2022,15, 3043 2 of 19
found everywhere in geological media, thereby making nanoconfinement effects essential
for the proper understanding of many phenomena in geoscience, including the physical
and mechanical properties of saturated porous media [
18
], formation of superhydrated
phases of minerals [
19
,
20
], dehydration processes of subducting slabs [
20
], deformation and
rupture of heterogeneous geomaterials [
21
], and lubrication effects under high-pressure
conditions [
22
,
23
]. Confinement [
22
] and nanoconfinement [
23
–
27
] strongly influence the
mechanical and frictional properties of lubricants in highly loaded contacts.
Taking into account nanoconfinement-related effects is important in solving many
materials science and nanotechnology problems, including the development of functional
nanomaterials (e.g., molecular sieves and ion-selective membranes [
28
–
33
]), bionanosen-
sors [
34
], highly ordered mesostructured composites with unusual mechanical and struc-
tural properties [
35
], auxetic materials [
36
], efficient sorbents [
37
], etc. [
38
–
40
]. Additionally,
in a variety of cases, nanoconfinement provides favorable conditions for the catalysis of
chemical reactions [41–43].
It is well known that confinement of thin layers has a strong effect on the phase state
and phase transformations of a substance. Water confined in slit-shaped nanopores of
7 Å width between two graphite plates shows solid-like behavior, whereas in pores of 9 Å
width or more this effect is not observed [
44
]. A similar effect (formation of 2D ice crystals)
was recently studied for water bilayers between mica and graphene [
45
,
46
]. Moreover,
the mechanism of nanoconfinement-induced freezing of liquids was experimentally in-
vestigated for cyclohexane, octamethylcyclotetrasiloxane, and toluene [
47
]. It was found
that when the nanopore width is below a certain critical value—about six molecular layers
of confined liquid—a liquid-like–solid-like phase transition occurs. Another example is
argon; when confined between atomically flat graphene nanosheets, argon does exist in
a solid phase, with mixed hexagonal close-packed (HCP) and face-centered cubic (FCC)
structures, at pressures significantly lower than the crystallization pressure of argon in a
bulk system [48].
Water is among the most interesting subjects for the study of nanoconfined matter, due
to its ubiquitous presence and importance for our planet. Even in a bulk state, water shows
an unusually large variety of structural forms (including many phases of ice [
49
]), and has
some well-known anomalous features. Confined water plays a key role in many practically
significant processes, such as the functioning of biological molecular machines [
50
], selective
ion filtration through biological channels [
51
], functioning of porins, and changes in the
properties of biological components in soft nanoconfinement [52].
The character of the interaction between water and the confining medium is largely
determined by the hydrophobicity/hydrophilicity of the pore walls which, in turn, depends
on both the atomic structure of the surface and the distribution of electric charge between
surface groups of atoms, i.e., surface polarity [53–55].
The present study is devoted to the question of how surface polarity can influence the
network structure and the density of hydrogen bonds in nanoconfined water, as well as
its mechanical properties at extremely high pressures. To carry out the analysis, we used
molecular dynamics (MD) simulation with atom-level models. We considered brucite-type
layered metal hydroxides as the nanopore wall material. This is a widespread natural
mineral class defined by the chemical composition Me
(II)
(OH)
2
, where Me
(II)
is a divalent
metal [
56
] or a mixture of metals [
57
]. The MD model allowed us to study the properties
and structure of water trapped in the interlayers of typical layered hydroxide minerals
under different pressures, and to directly compare the behavior of nanoconfined water
under hydrophilic and hydrophobic conditions in otherwise identical nanopores.
2. Materials and Methods
2.1. Molecular Dynamics Model
Molecular dynamics simulation with atomic-level description is a convenient tool for
studying matter under nanoconfinement. To investigate the structure and properties of
nanoconfined water, as well as the influence of wall–water electrostatic interaction on water
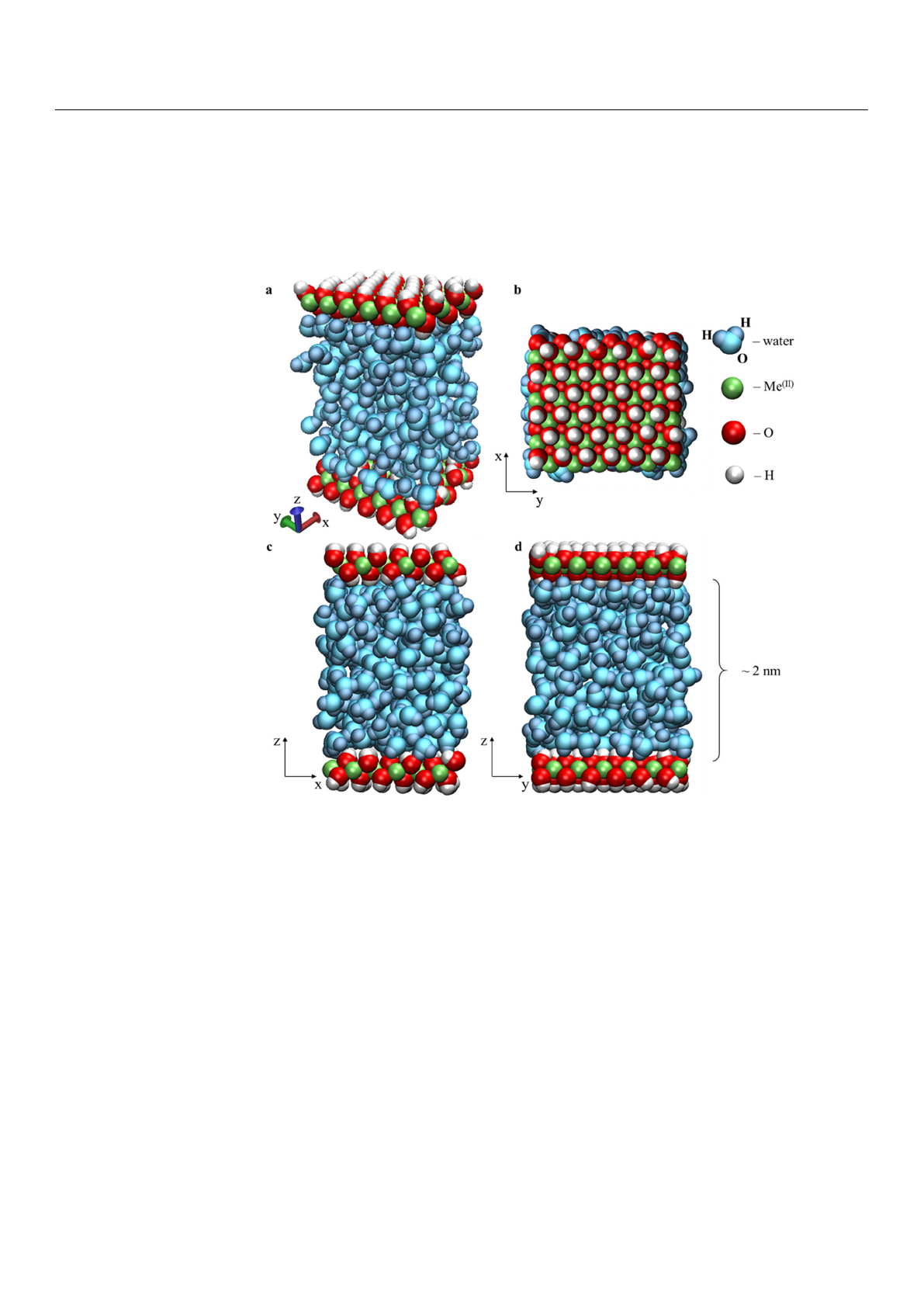
Materials 2022,15, 3043 3 of 19
behavior under high-pressure conditions, two molecular dynamics models of a slit-shaped
nanopore filled with water were developed. Each model consisted of an approximately
2 nm thick layer of water confined by two parallel metal hydroxide nanosheets located in
the x–y plane (Figure 1). Periodic boundary conditions (PBCs) were applied along the xand
yaxes.
Materials 2022, 15, x FOR PEER REVIEW 3 of 20
2. Materials and Methods
2.1. Molecular Dynamics Model
Molecular dynamics simulation with atomic-level description is a convenient tool
for studying matter under nanoconfinement. To investigate the structure and properties
of nanoconfined water, as well as the influence of wall–water electrostatic interaction on
water behavior under high-pressure conditions, two molecular dynamics models of a
slit-shaped nanopore filled with water were developed. Each model consisted of an ap-
proximately 2 nm thick layer of water confined by two parallel metal hydroxide
nanosheets located in the x–y plane (Figure 1). Periodic boundary conditions (PBCs) were
applied along the x and y axes.
Figure 1. Initial configuration of the modelled system’s periodic boxes 36[2Me
(II)
(OH)
2
·7H
2
O]
equilibrated at the pressure 0.1 GPa: perspective view (a), top view (b), x–z view (c), and y–z views
(d).
In the first model, the nanopore walls are representative of a class of naturally oc-
curring metal hydroxides Me
(II)
(OH)
2
. The structural parameters of divalent iron hy-
droxide Fe(OH)
2
with near-medial lattice constant a [56] were chosen as representative
parameters for layered hydroxides of divalent metals within the trigonal crystal system
(Supplementary Table S1 and Supplementary Figure S1).
The second model is equivalent to the first one, except for modified partial atomic
charges (PACs), whose values were reduced by a factor of 10 compared to the first model
(Supplementary Table S2). Henceforth, we will refer to the first model as the “Naturally
charged host model” and the second model as the “Low-charged host model”. It should
be noted that both models have the overall composition 36[2Me
(II)
(OH)
2
·7H
2
O], and that
there is no net electric charge. In other words, we studied two “host-guest” systems with
the same atomic structure of the nanopore walls and the same number of “guest” water
Figure 1.
Initial configuration of the modelled system’s periodic boxes 36[2Me
(II)
(OH)
2·
7H
2
O] equili-
brated at the pressure 0.1 GPa: perspective view (a), top view (b), x–z view (c), and y–z views (d).
In the first model, the nanopore walls are representative of a class of naturally occur-
ring metal hydroxides Me
(II)
(OH)
2
. The structural parameters of divalent iron hydroxide
Fe(OH)
2
with near-medial lattice constant a[
56
] were chosen as representative parameters
for layered hydroxides of divalent metals within the trigonal crystal system (Supplementary
Table S1 and Supplementary Figure S1).
The second model is equivalent to the first one, except for modified partial atomic
charges (PACs), whose values were reduced by a factor of 10 compared to the first model
(Supplementary Table S2). Henceforth, we will refer to the first model as the “Naturally
charged host model” and the second model as the “Low-charged host model”. It should
be noted that both models have the overall composition 36[2Me
(II)
(OH)
2·
7H
2
O], and that
there is no net electric charge. In other words, we studied two “host-guest” systems with
the same atomic structure of the nanopore walls and the same number of “guest” water
molecules. The only difference was that the confining walls were hydrophilic in the first
model, and hydrophobic in the second.
The crystal structure obtained for deuterated iron(II) hydroxide Fe[O(H
0.86
D
0.14
)]
2
by Parise et al. [
58
] was used for the nanosheet wall model. This material was chosen
because it has a crystal structure similar to many brucite-like metal hydroxides (such
as Ni(OH)
2
, Mg(OH)
2
, Co(OH)
2
, Zn(OH)
2
, Mn(OH)
2
, Cd(OH)
2
, Ca(OH)
2
), and has an

Materials 2022,15, 3043 4 of 19
intermediate lattice constant among them (see comparison in Supplementary Table S1 and
Supplementary Figure S1).
The force field parameters that describe the nanopore walls (including the PACs,
Lennard-Jones, and covalent bond parameters) were assigned in the naturally charged host
model in accordance with the CLAYFF force field [
59
]. The only exception was that the
Lennard-Jones parameters for hydrogen atoms were parameterized as described in [
60
],
with non-zero values r
0
= 0.449 Å and
ε
= 0.1925 kJ/mol [
61
], aiming for better compatibility
with the TIP3P water model. Partial atomic charges of walls in the low-charged host model
system were 10 times lower than in the first model. The PAC values used in the present
study are listed in Supplementary Table S2.
Both models contain a constant number of water molecules (N
w
= 7
×
36 = 252)
confined between two parallel nanosheets of metal hydroxide. Each nanosheet contains 36
Me
(II)
(OH)
2
units. The entire system can therefore be described as 36[2Me
(II)
(OH)
2·
7H
2
O].
The TIP3P model was used to parameterize the water molecules [
62
]. The covalent
bonds and angles of the model are described in Supplementary Figure S2 and Supplemen-
tary Table S3.
The dimensions of the simulation box—X = 16.965 Å and Y = 19.590 Å—remained
unchanged during the runs. The size of the system along the zaxis and, therefore, the
width of the nanopore, was self-adjusting depending on external load. The simulation for
each applied pressure was 3 ns long.
Larger systems were additionally prepared by replication of the base models twice
along both the xand yaxes. The dimensions of the enlarged system were X = 33.93 Å,
Y = 39.18 Å, with an overall formula 144[2Me
(II)
(OH)
2·
7H
2
O]. These models contained 1008
water molecules. They were used in the series of additional verifying simulations near the
phase transformation points. The duration of each additional simulation was 5 ns.
The cutoff distance for pairwise interactions was 10 Å, with switching to a smooth
damping function starting at 8 Å. The long-range term of the electrostatic interaction was
evaluated with the PPPM (particle–particle–particle–mesh) method, with a relative error of
10
−4
[
63
]. Temperature was constant and equal to 310 K in all simulations. The temperature
was maintained by a Nose–Hoover thermostat [64]. The time step was 1 fs.
The traditional approach in molecular dynamics to the implementation of confinement
of matter is the use of rigid [
65
] and elastic [
66
] walls. We used the rigid wall approximation,
with all heavy (non-hydrogen) atoms of the Me
(II)
(OH)
2
being part of a single rigid body
(one rigid body per nanosheet). Thus, the metal and oxygen atoms of the walls were able
to move only in the vertical direction z. Surface hydrogen atoms, being covalently bonded
with oxygen atoms, had no constraints applied.
The model samples were subjected to uniaxial compressional loading along the z-axis
(see Figure 1). The coordinates of the heavy atoms of the lower wall were fixed during
simulation, whereas the upper wall behaved as a piston, providing the specified load on
the confined water along the zaxis (Figure 1). The load was applied by an external force
-f
z
, which was uniformly distributed over all non-hydrogen atoms of the upper wall piston.
The force was directed downward; its magnitude was calculated as the product of the
specified pressure and the area of the wall–piston surface. The external (applied) pressure
pvaried in the range of 0.1–10 GPa. This range particularly corresponds to tectonic plates
at the characteristic depth interval from 3 km (upper crust) to 300 km (upper mantle) [20],
or to highly stressed contact spots in heavily loaded tribological couples [67].
Using the described models, a comprehensive analysis of nanoconfined water structure
and properties at different pressures was carried out. The analysis covered:
1. Water density and compressibility;
2.
Spatial arrangement of water molecules, including the radial distribution functions
(RDFs);
3. The number of hydrogen bonds;
4. The self-diffusion coefficient.

Materials 2022,15, 3043 5 of 19
2.2. Advantages of the Model
As previously mentioned, the choice of the Fe(OH)
2
structure with a lattice constant
of a= 3.265 Å [
58
] as the basis of the model allowed us to extend the results of the study to
the general class of brucite-like layered hydroxides of divalent metals, or at least to those
of Ni (3.117 Å), Mg (3.147 Å), Co (3.173 Å), Zn (3.194 Å), Mn (3.316 Å) and, probably, Cd
(3.499 Å), due to the similar values of their lattice constants [44].
2.3. Assumptions of the Model
We assumed that the utilized force field parameters were sufficiently accurate to model
the studied systems at high pressures, and in particular that the TIP3P model was suitable
for describing water behavior up to a pressure of 10 GPa
We also assumed that the mineral did not change its structure under loading and,
therefore, that the rigid model was applicable (except for surface hydrogen atoms). This
assumption is partially supported by data obtained by Hermann and Mookherjee [
68
].
According to a computationally derived thermodynamic phase diagram for brucite at
~300 K [
68
], the brucite Mg(OH)
2
retains its layered structure up to a pressure of ~20 GPa.
The rigid model of the wall structure is consistent with these data.
2.4. Limitations of the Model
The modeled systems had a fixed ratio of water molecules N
w
to the number of metal
atoms (7:2), and only the case of constant dimensions along the xand yaxes was considered.
3. Results
3.1. Structure and Properties of Nanoconfined Water
The simulations revealed two solid high-pressure phases of water confined between natu-
rally charged Me
(II)
(OH)
2
sheets (in addition to the low-pressure liquid phase). These phases can
be clearly seen in all numerically obtained dependencies (see Figures 2,3,4and 5a,c). Two phase
transitions at p
1
~3.1 GPa and p
2
~6.7 GPa were determined within the studied pressure range for
the base model 36[2Me
(II)
(OH)
2·
7H
2
O] (Figure 2a, black curves). More accurate estimates were
obtained using the fourfold-enlarged model 144[2Me
(II)
(OH)
2·
7H
2
O] in prolonged simulations
(Figure 4, red curves in the left and central panels): p1~3.3–3.4 GPa, and p2~6.7–7.1 GPa.
In the first phase transition at the pressure p
1
, the random arrangement of water
molecules became ordered (Figure 3a), and the diffusion coefficient of water decreased
by three orders of magnitude (Figure 5c, solid black curve) from D~3.24
×
10
−10
m
2
/s to
4.33
×
10
−13
m
2
/s. It is worth mentioning here that our estimate of the self-diffusion
coefficient at p= 0.1 GPa for nanoconfined water was 3.1
×
10
−9
m
2
/s, which is of the same
order of magnitude as that of bulk water Dbulk~2.299 ×10−9m2/s [69].
According to generally accepted data, ordinary water in the bulk state crystallizes
into ice VI [
70
] at a phase transition pressure of about 1.0–1.17 GPa (at temperatures of
300–310 K) (see Supplementary Equations (1)–(3)). However, experimental evidence of the
possibility of metastable supercompressed bulk liquid water at higher pressures has also
been reported [
71
]. Dolan et al. [
72
] were the first to show the formation of cubic ice (ice
VII) under shock compression with GPa peak pressures. They particularly showed that the
solid phase becomes more stable than the liquid phase at applied pressures above 2 GPa,
and that the phase transformation takes place within nanoseconds instead of seconds
under ambient conditions. Later, Dolan et al. [
73
] demonstrated a practical limit for the
metastable liquid phase. They showed that bulk water under isentropic shock compression
can homogeneously transform into a high-pressure phase (ice VII) at a pressure of 7 GPa
(a practical limit for the metastable liquid phase) [
73
–
75
]. The abovementioned values
1 GPa and 7 GPa bound the pressure range in which crystallization of water is possible
at ambient or moderately elevated temperatures. The specific value of crystallization
pressure depends (at least for microscale, and even more so for nanoscale volumes of water)
on interface hydrophilicity—that is, on hydrogen bonding between the wall surface and
water [73,74].
Loading more pages...