
Citation: Krebs, S.K.; Stech, M.; Jorde,
F.; Rakotoarinoro, N.; Ramm, F.;
Marinoff, S.; Bahrke, S.; Danielczyk,
A.; Wüstenhagen, D.A.; Kubick, S.
Synthesis of an Anti-CD7
Recombinant Immunotoxin Based on
PE24 in CHO and E. coli Cell-Free
Systems. Int. J. Mol. Sci. 2022,23,
13697. https://doi.org/10.3390/
ijms232213697
Academic Editor: Antonio Maccio
Received: 19 October 2022
Accepted: 5 November 2022
Published: 8 November 2022
Publisher’s Note: MDPI stays neutral
with regard to jurisdictional claims in
published maps and institutional affil-
iations.
Copyright: © 2022 by the authors.
Licensee MDPI, Basel, Switzerland.
This article is an open access article
distributed under the terms and
conditions of the Creative Commons
Attribution (CC BY) license (https://
creativecommons.org/licenses/by/
4.0/).
International Journal of
Molecular Sciences
Article
Synthesis of an Anti-CD7 Recombinant Immunotoxin Based on
PE24 in CHO and E. coli Cell-Free Systems
Simon K. Krebs 1,2 , Marlitt Stech 1, Felix Jorde 1, Nathanaël Rakotoarinoro 1,3, Franziska Ramm 1,4,
Sophie Marinoff 5, Sven Bahrke 5, Antje Danielczyk 5, Doreen A. Wüstenhagen 1and Stefan Kubick 1,4,6,*
1
Branch Bioanalytics and Bioprocesses (IZI-BB), Fraunhofer Institute for Cell Therapy and Immunology (IZI),
Am Mühlenberg 13, 14476 Potsdam, Germany
2Institute for Biotechnology, Technical University of Berlin, Ackerstrasse 76, 13355 Berlin, Germany
3Institute of Pharmacy, Freie Universität Berlin, Königin-Luise-Strasse 2 + 4, 14195 Berlin, Germany
4Institute of Chemistry and Biochemistry, Freie Universität Berlin, Takustrasse 6, 14195 Berlin, Germany
5Glycotope GmbH, Robert-Roessle-Strasse 10, 13125 Berlin, Germany
6Faculty of Health Sciences, Joint Faculty of the Brandenburg University of Technology Cottbus-Senftenberg,
The Brandenburg Medical School Theodor Fontane and the University of Potsdam, 14476 Potsdam, Germany
*Correspondence: [email protected].de
Abstract:
Recombinant immunotoxins (RITs) are an effective class of agents for targeted therapy in
cancer treatment. In this article, we demonstrate the straight-forward production and testing of an
anti-CD7 RIT based on PE24 in a prokaryotic and a eukaryotic cell-free system. The prokaryotic
cell-free system was derived from Escherichia coli BL21 Star
TM
(DE3) cells transformed with a plasmid
encoding the chaperones groEL/groES. The eukaryotic cell-free system was prepared from Chinese
hamster ovary (CHO) cells that leave intact endoplasmic reticulum-derived microsomes in the cell-
free reaction mix from which the RIT was extracted. The investigated RIT was built by fusing an
anti-CD7 single-chain variable fragment (scFv) with the toxin domain PE24, a shortened variant of
Pseudomonas Exotoxin A. The RIT was produced in both cell-free systems and tested for antigen
binding against CD7 and cell killing on CD7-positive Jurkat, HSB-2, and ALL-SIL cells. CD7-positive
cells were effectively killed by the anti-CD7 scFv-PE24 RIT with an IC
50
value of 15 pM to 40 pM for
CHO and 42 pM to 156 pM for E. coli cell-free-produced RIT. CD7-negative Raji cells were unaffected
by the RIT. Toxin and antibody domain alone did not show cytotoxic effects on either CD7-positive
or CD7-negative cells. To our knowledge, this report describes the production of an active RIT in
E. coli and CHO cell-free systems for the first time. We provide the proof-of-concept that cell-free
protein synthesis allows for on-demand testing of antibody–toxin conjugate activity in a time-efficient
workflow without cell lysis or purification required.
Keywords:
cell-free protein synthesis; CFPS; cell-free expression; CFE; RIT; Pseudomonas Exotoxin
A; GroEL/GroES; DnaK/DnaJ/GrpE; trigger factor
1. Introduction
Cancer kills millions of people each year despite impressive achievements in oncology
during the last 100 years. Currently, most cancer cases are initially treated by classic chemo-
and radiotherapy regimens. More recently, personalized targeted therapy is increasingly
being used as second-line and third-line treatment options and, for some indications,
emerged as a first-line treatment. Examples for targeted therapy first-line drugs include
anti-Her2/neu monoclonal antibody Trastuzumab, BRAF kinase inhibitor Vemurafenib,
and anti-IL2 immunotoxin tagraxofusp. Targeted cancer therapy is potentially superior to
systemically effective cytotoxic drugs due to off-target effects and the concurrent narrow
therapeutic window of undirected anti-cancer cytotoxins. Survival rates of most malignan-
cies improved drastically over the last 100 years due to new drugs and improved regimens
Int. J. Mol. Sci. 2022,23, 13697. https://doi.org/10.3390/ijms232213697 https://www.mdpi.com/journal/ijms

Int. J. Mol. Sci. 2022,23, 13697 2 of 24
but many types of cancer are still highly lethal and cannot be cured with the drugs approved
for clinical use. This demonstrates the need for further treatment options [1–4].
A promising class of targeted anti-cancer therapeutics are antibody–toxin conjugates,
such as antibody–drug conjugates (ADCs) and recombinant immunotoxins (RITs). They
target a cancer-associated antigen and the attached toxin moiety specifically eradicates
malignant cells, while healthy cells remain unaffected. Thereby, the therapeutic window is
enlarged [
5
,
6
]. In contrast to ADCs, the cytotoxic compound in RITs is a protein, which in
most cases is of non-human origin. Prominent candidates for toxin domains include recom-
binant variants of the bacterial toxins Pseudomonas Exotoxin A (PE) and diphtheria toxin
(DT), which both target eukaryotic elongation factor 2 (eEF2). Furthermore, plant-derived
toxins, such as Ricin and Gelonin, are frequently used, which both target eukaryotic 28S
ribosomal RNA [
7
]. The associated cell killing triggered by translation inhibition and ac-
cording downstream effectors therefore exclusively affects eukaryotic cells [
8
]. Accordingly,
RIT production with toxin domains, which target the eukaryotic translation process, is
challenging in eukaryotic expression hosts due to self-intoxication effects [
9
,
10
]. To avoid
self-intoxication, prokaryotic expression hosts, such as Escherichia coli, are utilized. The
RITs are then usually purified from inclusion bodies (IBs) [
11
], which represents a time-
intensive procedure with relatively low efficiency [
12
,
13
]. In fact, the three FDA approved
RITs, denileukin diftitox (DT-IL-2, DAB389IL-2; Ontak
®
), tagraxofusp-erzs (DT-IL-3, SL-
401, Elzonris
®
), and moxetumomab pasudotox (anti-CD22 dsFv-PE38, CAT-8015 or HA22,
Lumoxiti®), are produced in E. coli cells and then refolded and purified from IBs [14–17].
Immunotoxins suffer from several drawbacks that need to be overcome before fully
unfolding their clinical potential. These critical issues concern all domains of the RIT,
namely targeting domain, linker domain, and toxin domain. For example, the immuno-
genicity of non-human antibodies and toxins leads to respective neutralizing antibodies
that limit the RIT’s efficacy. Furthermore, several potentially life-threatening conditions
evoked by off-target or on-target effects were observed in patients treated with RITs, such
as capillary leak syndrome, cytokine release syndrome, hepatotoxicity, nephrotoxicity, and
cardiac toxicity [
17
,
18
]. To overcome these issues and gain more effective RITs, iterative
redesign of the RIT coding sequence has to be undertaken, such as humanization of the
antibody, affinity maturation of complementarity determining regions, conversion to a
bispecific format, improvement of linker stability, linker functionality, and/or removal of
B and T cell epitopes of the toxin domain [
8
,
17
,
19
–
23
]. We anticipate that cell-free protein
synthesis (CFPS) may be applied as an economic and time-efficient tool to evaluate the
impact of sequence redesign on RIT efficacy during its development process. CFPS enables
on-demand production of proteins of interest (POIs) within a few hours, using the protein
translation machinery of disintegrated cells. The option for massive parallelization using
linear expression templates and the open nature of cell-free systems allows economic,
flexible, high-throughput screening of POIs on an analytical scale without the need to create
genetically modified organisms or to perform cell lysis of the expression host or purification
of the POI [24,25].
In this study, we explored E. coli and Chinese hamster ovary (CHO)-based CFPS as
a tool for straight-forward RIT production and
in vitro
testing for antigen binding and
cytotoxicity. We aimed to streamline the RIT development process by avoiding cumber-
some IB solubilization and purification to allow flexible on-demand synthesis of RITs.
The RIT chosen for this proof-of-concept targets CD7 by a single-chain variable fragment
(scFv) domain, which was linked to PE24 by a flexible GS linker. The toxin domain PE24
(also known as PE[LR]) has been developed by Pastan and colleagues [
26
]. Compared to
full-length PE (also known as PE66), in PE24, the native CD91 receptor binding domain
Ia and the translocation domain II, except the furin cleavage site, are deleted. PE24 is the
shortest version of PE66 currently known to effectively kill target cells when equipped
with an appropriate binding domain [
27
]. CD7, the RIT’s target, is a 40 kDa glycosy-
lated membrane protein mainly expressed on healthy T and NK cells including their
progenitors and is involved in T cell activation and immune cell interaction [
28
]. Several

Int. J. Mol. Sci. 2022,23, 13697 3 of 24
lymphatic diseases are associated with CD7-positive T cells or B cells, such as most cases of
T cell acute lymphoblastic leukemia (T-ALL) [
29
], some cases of acute myeloid leukemia
(AML) [
30
], and acute B lymphoblastic leukemia (B-ALL) [
31
], as well as graft vs. host
disease (GvHD) [
32
,
33
]. Promising treatment options have recently been approved by
the FDA for B-ALL and AML [
34
,
35
]. Potential indications for an anti-CD7 RIT include
relapsed or refractory T-ALL [
36
,
37
] and steroid-refractory acute GvHD [
38
,
39
] as well as
other treatments that require T cell depletion, such as allogeneic hematopoietic stem cell
transplantation (HSCT) [
40
,
41
]. However, CD7-targeted T cell depletion renders the patient
immune-deficient and therefore is associated with great risks for opportunistic infections
during treatment [37].
CHO cells are the most commonly used expression hosts for production of therapeutic
proteins, particularly for therapeutic antibodies [
42
]. This is mainly due to their ability for
complex post-translational modifications, such as glycosylation, and their recognition as a
safe expression host by the FDA [
43
]. However, due to self-intoxication, CHO cells are not
a viable option for production of RITs, whose toxin domain targets eukaryotic translation.
Therefore, to avoid work-intensive IB solubilization and purification associated with E. coli
cell-based expression, an E. coli cell-free system was chosen for RIT production. It is consid-
ered the most productive cell-free system [
44
]. In order to increase the soluble yield of the
RIT, the E. coli cell-free system was supplemented with the chaperones GroEL/GroES (ELS),
DnaK/DnaJ/GrpE (KJE), or Trigger Factor (TF) in different combinations. Chaperones
are indispensable, evolutionary, conserved folding helpers required to maintain protein
homeostasis in cells [
45
,
46
]. Supplementation of chaperones was found to often increase
soluble yields and activity of the POI by assisting correct folding of the heterologously
expressed POI in E. coli cell-based and in E. coli cell-free expression [47,48].
Aiming to provide a solution for endotoxin-free RIT production in a eukaryotic system,
we further evaluated CHO-based CFPS for RIT production. The CHO cell-free system used
here contains endoplasmic reticulum (ER)-derived microsomes. We therefore speculated
that CHO-based CFPS might be less susceptible to PE-mediated translation inhibition
than eukaryotic cell-based production. Microsomes are vesicular structures to which cell-
free synthesized proteins can be co-translationally targeted, e.g., by the signal peptide
from the bee toxin melittin [
49
]. Microsome-translocated proteins are physically separated
from the translation apparatus by the lipid bilayer of the microsomes. Accordingly, eEF2
would be protected from PE24-mediated ribosylation. Importantly, microsome-extracted
proteins most often show higher activity than proteins that have not been translocated.
This is likely due to an environment that is beneficial for correct folding, such as disulfide
bond promoting redox potential and ER-residing chaperones [
50
,
51
]. Furthermore, post-
translational modifications, such as core glycosylation and disulfide bond formation, can
be achieved in microsomes [49,50,52].
To our knowledge, neither CHO nor E. coli CFPS have ever been evaluated for RIT
production. Here, we demonstrate the proof-of-concept for RIT
in vitro
testing using a
CHO-based cell-free system containing ER-derived microsomes and an E. coli-based cell-
free system supplemented with ELS using an anti-CD7 scFv-PE24 RIT as a model. Using
cell-free systems for on-demand synthesis of RITs might help to streamline RIT research
and development.
2. Results
2.1. E. coli-Based Cell-Free Production of PE24 RIT
RITs are commonly produced using E. coli cell-based expression with the subsequent
purification and refolding of IBs [
17
]. Using a productive, well-established E. coli expression
strain as a basis for a cell-free system, we seek practical, economic, and time-saving advan-
tages when using CFPS for RIT screening purposes instead of E. coli cell-based production.
In this study, we evaluated an E. coli-based cell-free system for the production and testing
of the anti-CD7 model RIT. The E. coli cell-free system was equipped with chaperones
ELS, KJE, and TF [
47
] in different combinations by transformation of the E. coli strain

Int. J. Mol. Sci. 2022,23, 13697 4 of 24
BL21 Star
TM
(DE3) with the corresponding plasmid prior to lysate preparation. Soluble
yield and functionality of the scFv and the scFv-PE24 RIT synthesized in presence and
absence of chaperones were compared in order to identify the most suitable chaperone for
E. coli cell-free RIT production. Liquid scintillation counting revealed that, for the cell-free
system derived from the E. coli strain without chaperones, less than 20% of the total protein
(translation mix, TM) was found in the soluble fraction (supernatant, SN). In contrast, the
presence of chaperones substantially produced 2.5-fold to 8-fold increased yields in the
soluble fractions (Figure 1A,D). For scFv synthesis, the highest soluble yield was found
for the cell-free system prepared from the strain transformed with the plasmid coding
for ELS. For RIT synthesis, cell-free systems supplemented with KJE+ELS, ELS, or KJE
equally performed regarding yield in the soluble fraction, while the lowest increase in
soluble yield was found for the cell-free system equipped with TF+ELS. Visualization of the
synthesized proteins by autoradiography showed distinct signals at the expected molecular
weight. In addition, weaker signals at lower molecular weights were detected (Figure 1B,E).
Faint signals were furthermore detected in the autoradiograph of the no-template control
(NTC), indicating minor amounts of residual endogenous mRNA present in the lysate
(Figure S1A). In the corresponding blue gels, the overexpressed chaperones could be de-
tected in contrast to BL21 Star
TM
(DE3) wildtype without chaperones (Figure S1B–D). To
assess the impact of the chaperones on cell-free-produced scFv functionality, an indirect
eight-point enzyme-linked immunosorbent assay (ELISA) against CD7 with unpurified
SN was performed (Figure 1C). The IC
50
values of the scFv synthesized in the different
E. coli cell-free systems were in a similar range within one log. IC
50
values determined were
1.40 nM for wildtype BL21 Star
TM
(DE3), 1.91 nM for KJE+ELS, 1.24 nM for ELS, 8.38 nM
for KJE, and 2.76 nM for TF+ELS. As expected, the NTC did not demonstrate binding
(Figure S2). Furthermore, to determine the chaperones’ influence on the cytotoxicity of the
RIT, an 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide tetrazolium (MTT)
assay on CD7-positive Jurkat cells was performed (Figure 1F). For each sample and the
corresponding NTC, a 10-point dilution series was directly prepared from unpurified SN.
The absorbance value of the dilution of each sample was normalized for the absorbance
of the NTC value at the respective dilution, giving the percentage of surviving cells in
relation to NTC-treated cells. This allowed us to mathematically compensate for growth
inhibitory effects evoked by the addition of highly concentrated unpurified SN fraction
to cells (Figure S3A,B). The MTT assay revealed comparable IC
50
values with 176 pM for
BL21 without chaperones, 172 pM for KJE+ELS, 139 pM for ELS, 310 pM for KJE, and
901 pM for TF+ELS. We noted that ELS produced the highest soluble yield and slightly
increased activity for scFv and RIT compared to the other E. coli cell-free systems. Based
on the data, we decided to proceed for the following experiments with the E. coli cell-free
system equipped with ELS.
The scFv, PE24, and the RIT were synthesized in the E. coli cell-free system with ELS
chaperones and the soluble fraction (SN) was prepared (Figure 2A). Liquid scintillation
counting determined the yield of soluble protein to be between 45 and 82
µ
g/mL (Figure 2B),
which was comparable to scFv and RIT yield obtained for the ELS supplemented cell-
free system in the chaperone screening (Figure 1A,D). The POIs showed their expected
molecular weight in the autoradiograph and weaker signals at lower molecular weights
(Figure 2C). In an indirect 8-point anti-CD7 ELISA with unpurified SN, an IC
50
value of
1.07 nM was calculated for the scFv binding to CD7 (Figure 2D). The determined IC
50
value
was in positive agreement with the data for ELS-supplemented CFPS in the chaperone
screening (Figure 1C). The RIT bound to CD7 with an IC
50
value of 2.01 nM, while for PE24,
no binding was detected. The NTC did not show any binding either (Figure S4).
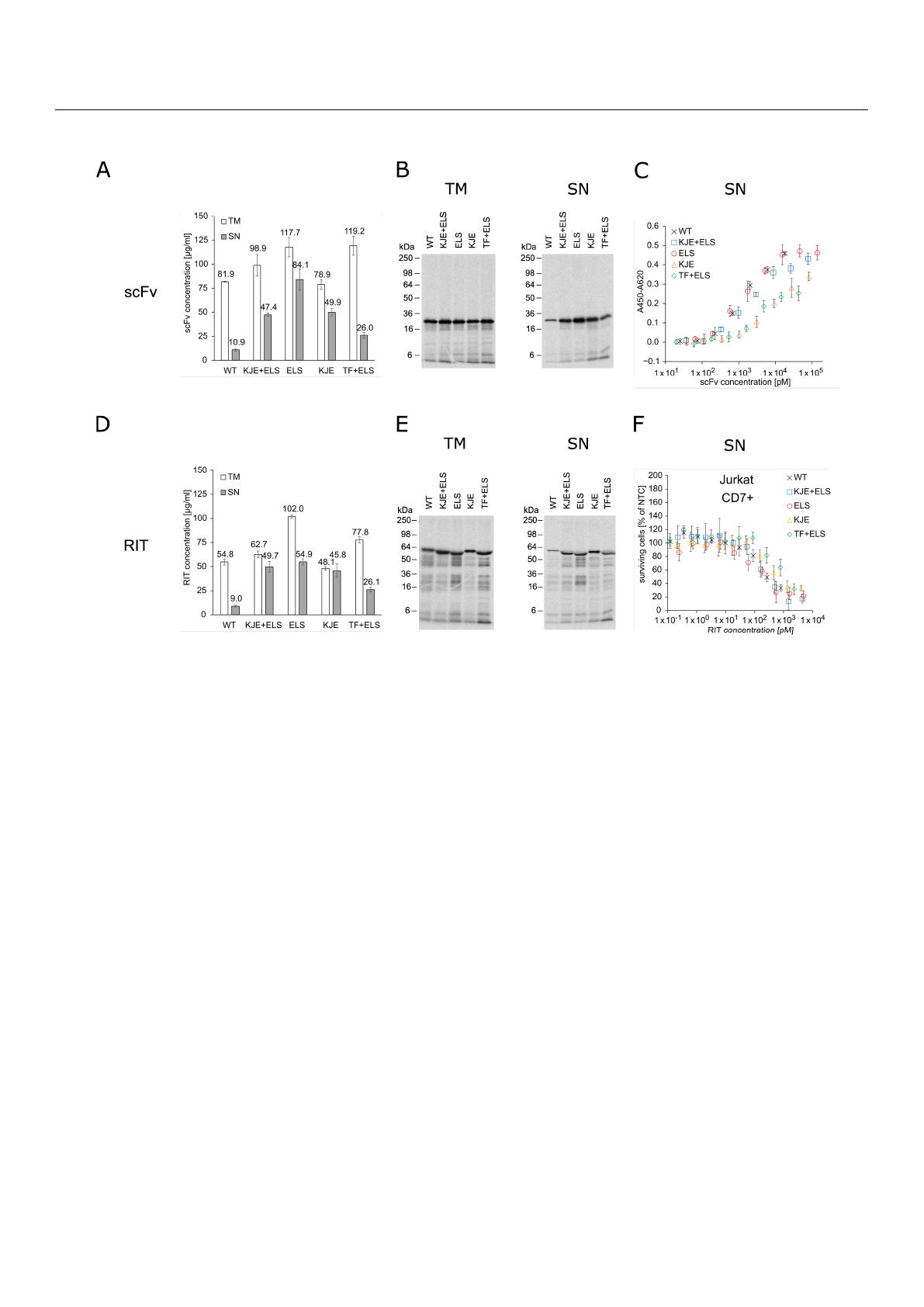
Int. J. Mol. Sci. 2022,23, 13697 5 of 24
Int. J. Mol. Sci. 2022, 23, x FOR PEER REVIEW 5 of 25
Figure 1. Quantification, autoradiography, antigen binding, and cytotoxicity of E. coli cell-free-pro-
duced proteins with and without chaperones. (A) Liquid scintillation counting for quantification of
scFv in TM and SN fraction. Error bars represent the standard deviation of triplicate analysis. (B)
Autoradiography of reducing denatured 12% Tris-Glycine SDS-PAGE gel of scFv (29.9 kDa) in TM
and SN fraction. (C) Indirect ELISA to analyze binding of the scFv to its target antigen CD7. Ab-
sorbance values in wells without CD7 were subtracted from absorbance values in wells coated with
CD7. Error bars represent the standard deviation of triplicate analysis. (D) Liquid scintillation
counting for quantification of RIT in TM and SN fraction. Error bars represent the standard devia-
tion of triplicate analysis. (E) Autoradiography of reducing denatured 12% Tris-Glycine SDS-PAGE
of RIT (56.4 kDa) in TM and SN fraction. (F) MTT assay of RIT on CD7-positive Jurkat cells. Absorb-
ance values of samples were normalized in respect to absorbance of no-template control (NTC),
which was identically diluted and treated as the sample. Error bars represent the standard deviation
of normalized cell survival performed in triplicate. Abbreviations: scFv: single-chain variable frag-
ment; RIT: recombinant immunotoxin; TM: translation mix; SN: supernatant; WT: wildtype BL21
StarTM (DE3) w/o chaperones; KJE+ELS: WT transformed with plasmid coding for
DnaK/DnaJ/GrpE and GroEL/GroES; ELS: WT transformed with plasmid coding for GroEL/GroES;
KJE: WT transformed with plasmid coding for DnaK/DnaJ/GrpE; TF+ELS: WT transformed with
plasmid coding for Trigger Factor and GroEL/GroES.
The scFv, PE24, and the RIT were synthesized in the E. coli cell-free system with ELS
chaperones and the soluble fraction (SN) was prepared (Figure 2A). Liquid scintillation
counting determined the yield of soluble protein to be between 45 and 82 µg/mL (Figure
2B), which was comparable to scFv and RIT yield obtained for the ELS supplemented cell-
free system in the chaperone screening (Figure 1A,D). The POIs showed their expected
molecular weight in the autoradiograph and weaker signals at lower molecular weights
(Figure 2C). In an indirect 8-point anti-CD7 ELISA with unpurified SN, an IC50 value of
1.07 nM was calculated for the scFv binding to CD7 (Figure 2D). The determined IC50 value
was in positive agreement with the data for ELS-supplemented CFPS in the chaperone
screening (Figure 1C). The RIT bound to CD7 with an IC50 value of 2.01 nM, while for
PE24, no binding was detected. The NTC did not show any binding either (Figure S4).
Figure 1.
Quantification, autoradiography, antigen binding, and cytotoxicity of E. coli cell-free-
produced proteins with and without chaperones. (
A
) Liquid scintillation counting for quantification
of scFv in TM and SN fraction. Error bars represent the standard deviation of triplicate analysis.
(
B
) Autoradiography of reducing denatured 12% Tris-Glycine SDS-PAGE gel of scFv (29.9 kDa) in
TM and SN fraction. (
C
) Indirect ELISA to analyze binding of the scFv to its target antigen CD7.
Absorbance values in wells without CD7 were subtracted from absorbance values in wells coated
with CD7. Error bars represent the standard deviation of triplicate analysis. (
D
) Liquid scintillation
counting for quantification of RIT in TM and SN fraction. Error bars represent the standard deviation
of triplicate analysis. (
E
) Autoradiography of reducing denatured 12% Tris-Glycine SDS-PAGE of RIT
(56.4 kDa) in TM and SN fraction. (
F
) MTT assay of RIT on CD7-positive Jurkat cells. Absorbance
values of samples were normalized in respect to absorbance of no-template control (NTC), which
was identically diluted and treated as the sample. Error bars represent the standard deviation of
normalized cell survival performed in triplicate. Abbreviations: scFv: single-chain variable fragment;
RIT: recombinant immunotoxin; TM: translation mix; SN: supernatant; WT: wildtype BL21 StarTM
(DE3) w/o chaperones; KJE+ELS: WT transformed with plasmid coding for DnaK/DnaJ/GrpE and
GroEL/GroES; ELS: WT transformed with plasmid coding for GroEL/GroES; KJE: WT transformed
with plasmid coding for DnaK/DnaJ/GrpE; TF+ELS: WT transformed with plasmid coding for
Trigger Factor and GroEL/GroES.
Loading more pages...