
Cycloaddition
Silylium-Ion-Promoted (5++1) Cycloaddition of Aryl-Substituted
Vinylcyclopropanes and HydrosilanesInvolving Aryl Migration
TaoHe, Guoqiang Wang,Vittorio Bonetti, Hendrik F. T. Klare,and Martin Oestreich*
In memory of Professor Kilian MuÇiz (1970–2020)
Abstract: Atransition-metal-free (5++1) cycloaddition of aryl-
substituted vinylcyclopropanes (VCPs) and hydrosilanes to
affordsilacyclohexanes is reported. Catalytic amounts of the
trityl cation initiate the reaction by hydride abstraction from the
hydrosilane,and further progress of the reaction is maintained
by self-regeneration of the silylium ions.The new reaction
involves a[1,2] migration of an aryl group,eventually furnish-
ing 4- rather than 3-aryl-substituted silacyclohexane derivatives
as major products.Various control experiments and quantum-
chemical calculations support amechanistic picture where
asilylium ion intramolecularly stabilized by acyclopropane
ring can either undergo akinetically favored concerted
[1,2] aryl migration/ring expansion or engage in acyclopro-
pane-to-cyclopropane rearrangement.
Introduction
There is arich chemistry associated with substituted
vinylcyclopropanes (VCPs), especially because of their value
as C5 synthons for the construction of complex carbon
skeletons.[1] VCPs engage in adiverse set of bond reorganiza-
tions,[2] and transition-metal-catalyzed cycloadditions of
VCPs continue to attract considerable attention.[3] Aside
from those exciting synthetic applications,the parent VCP
1aroused interest in carbocation chemistry as its protonation
allowed the study of the stabilizing effect of the cyclopropyl
group on carbenium ions.[4] Thecyclopropylcarbinyl cation 2
is known to undergo various rearrangements (Scheme 1, top
left),[2,4] and we asked ourselves what would happen upon
treatment of 1with asilylium ion instead of astrong Brønsted
acid (Scheme 1, top right). Theanalogy lies in FlemingQsearly
notion of silylium ions being fat protons,[5] and the result
would likely be the carbenium ion 3further stabilized by the
b-silicon effect.[6] Assuming that the cyclopropane ring would
not be directly opened by the silylium ion,[7] the fate of 3
would not be predictable,and we expected new chemistry to
emerge.The plan was to initiate the reactions of the VCPs 4
and dihydrosilanes 5with catalytic amounts of trityl borate
Ph3C+[B(C6F5)4]@,relying on hydride abstraction (Corey
reaction[8])and, as such, self-regeneration of silylium
ions.[7,9,10] Thus,carbenium ion intermediates such as 6[11]
would be captured by hydride.Wechose the aryl-substituted
VCPs 4as model substrates and found several cyclic silanes
(7—9)asproducts (Scheme 1, bottom) but no simple alkene
hydrosilylation (4!10).[12] Thesix-membered-ring compound
7is the result of aformal (5++1) cycloaddition accompanied by
an unexpected migration of the aryl group.Wepresent here
the scope of the new reaction and its experimental and
quantum-chemical mechanistic analysis.
Results and Discussion
Using catalytic amounts of Ph3C+[B(C6F5)4]@as an
initiator,webegan our investigation with the reaction of 4a
and excess Et2SiH2(5a)inbenzene at ambient temperature
(Table 1). Themajor product was the 4-phenyl-1-silacyclo-
hexane 7aa along with small quantities of the 3-phenyl-1-
Scheme 1. Cyclopropyl-substitutedcarbenium ions and their potential
intermediacy in asilylium-ion-promoted (5++1) cycloaddition.
[*] Dr.T.He, Dr.G.Wang, V. Bonetti, Dr.H.F.T.Klare,
Prof. Dr.M.Oestreich
Institut ffrChemie, Technische Universit-tBerlin
Strasse des 17. Juni 115, 10623 Berlin (Germany)
E-mail:m[email protected]
Homepage:http://www.organometallics.tu-berlin.de
Supportinginformation and the ORCID identification number(s) for
the author(s) of this article can be found under:
https://doi.org/10.1002/anie.202004320.
T2020 The Authors. Published by Wiley-VCH Verlag GmbH &Co.
KGaA. This is an open access article under the terms of the Creative
Commons Attribution Non-Commercial License, which permits use,
distribution and reproduction in any medium, provided the original
work is properly cited, and is not used for commercial purposes.
A
ngewandte
Chemie
Research Articles
How to cite: Angew.Chem. Int. Ed. 2020,59,12186–12191
International Edition: doi.org/10.1002/anie.202004320
German Edition: doi.org/10.1002/ange.202004320
12186 T2020 The Authors. Published by Wiley-VCH Verlag GmbH &Co. KGaA, Weinheim Angew.Chem.Int. Ed. 2020,59,12186 –12191
silacyclohexane 8aa and 3-benzyltetrahydrosilole 9aa (en-
try 1). Thearene solvent influenced the product distribution,
and the formation of 9aawas reduced in 1,2-dichlorobenzene
and chlorobenzene (entries 2and 3). Proceeding with chlor-
obenzene,welooked into the variation of other parameters
(entries 4–6). Neither alower catalyst loading (1.0 instead of
2.0 mol%) nor less dihydrosilane (5.0 to 1.5 equiv) had an
effect on the reaction outcome.Aslight decrease in yield and
asomewhat less favorable product ratio was detected with
equimolar quantities of the reactants (entry 7). Thesix-
membered 7aa did form in 90%yield under the optimized
reaction conditions.
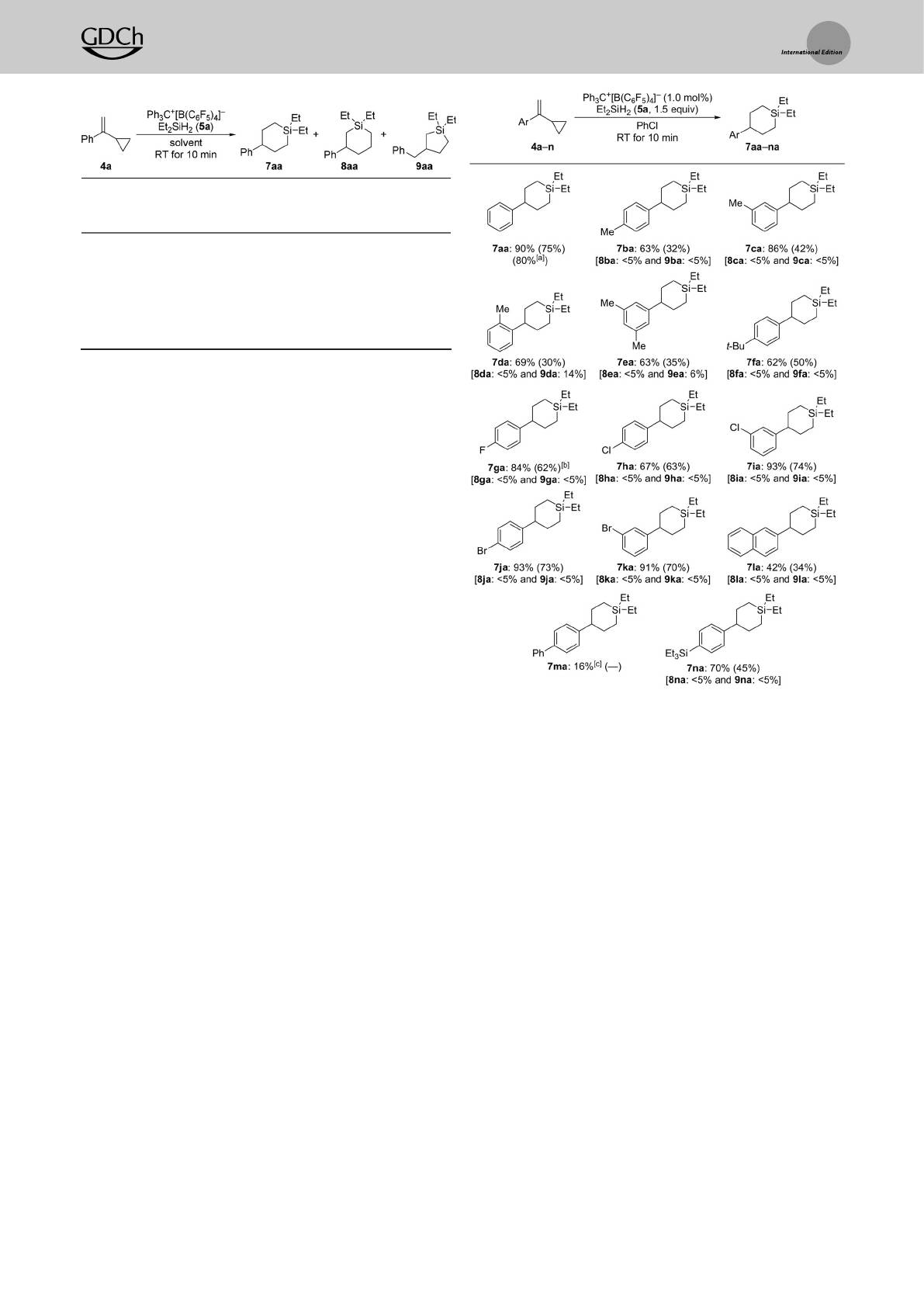
We then probed the substrate scope under the optimized
protocol (Scheme 2). Electronic and steric modifications of
the substituent on the aryl group were examined with 4a–k.
Theparent VCP 4ayielded 7aain 75%yield on a0.25 mmol
scale and in 80%yield on a7.0 mmol scale.Substrates
bearing,for example,electron-donating 4-methyl (4b), 3,5-
dimethyl (4e), and 4-tert-butyl (4f)groups reacted smoothly,
affording the desired products 7ba,7ea,and 7fa,respectively,
in moderate yields.Itisworthy of note that amethyl group in
the ortho position, as in 4d,did not affect the reactivity
compared with 4band 4c.Halogen atoms were tolerated well
in this reaction, as shown for the cases with fluorine (4g),
chlorine (4hand 4i), and bromine (4jand 4k)inthe para or
meta positions.The preparation of the ortho-substituted
regioisomers had failed. Abulkier b-naphthyl group as in 4l
was also compatible with the reaction conditions although
alower yield was obtained. Conversely,the biphenyl-sub-
stituted 4m hardly converted into the desired product 7ma.
We believe that the aryl substituents in 4l and 4m are more
likely to engage in electrophilic aromatic substitution with
silylium-ion intermediates,[13] thereby consuming the silylium
ion to result in either decomposition or low conversion. A
triethylsilyl group,asin4n,did not interfere with the (5++1)
cycloaddition. Theyields of the isolated products were
generally low because of the challenging purification of these
nonpolar compounds;yields refer to analytically pure mate-
rial and come close to those determined by 1HNMR
spectroscopy with less rigor.Wealso note here that the
reaction of aVCP with acyclohexyl, instead of the phenyl
group,led to acomplex reaction mixture (not shown).
Aside from the dihydrosilane used above,weasked
ourselves whether tertiary hydrosilanes would also participate
in this reaction. We had recently shown that silylium ions can
indeed cleave Si@C(sp3)bonds.[14] This dealkylation corre-
sponds to an exchange of an alkyl group between aquaternary
silane and asilylium ion. Therefore,intermediates with no Si@
Hbond available anymore could potentially still engage in the
Table 1: Optimization of the trityl-cation-initiated (5++1) cycloaddition.[a]
Entry Initiator
(mol%)
Dihydrosilane
(equiv)
Solvent Yield [%][b]
7aa8aa 9aa
12.0 5.0 PhH 80 <511
22.0 5.0 o-C6H4Cl281 <58
32.0 5.0 PhCl 90 <56
41.0 5.0 PhCl 90 <56
51.0 3.0 PhCl 90 <55
61.0 1.5 PhCl 90 <55
71.0 1.0 PhCl 75 95
[a] All reactions were performed with 4a (0.25 mmol) and the indicated
amounts of the initiator Ph3C+[B(C6F5)4]@and Et2SiH2(5a)under argon
atmosphere in the indicated arene solvent (2.5 mL, 0.1m)atroom
temperature. Conversion was greater than 95%for each entry as
determined by 1HNMR spectroscopy using CH2Br2as an internal
standard. [b] Yields determined by 1HNMR spectroscopy using CH2Br2
as an internalstandard.
Scheme 2. Scope I: Variation of the aryl group in the VCPs 4in the
(5++1) cycloadditonwith Et2SiH2.Unless otherwise noted, all reactions
were performed on 0.25 mmol scale, and conversion was greater than
95%for each entry.Conversions and yields were determined by
1HNMR spectroscopy using CH2Br2as an internalstandard. Yields
determined by 1HNMR spectroscopy using CH2Br2as an internal
standard. Yields of analytically pure material obtained after flash
chromatography on silica gel are given within parentheses. [a] Yield of
isolated product on a7.0 mmol scale. [b] Ph3C+[B(C6F5)4]@(2.0 mol%)
and Et2SiH2(5.0 equiv) were used. [c] 82% 4m was recovered.
A
ngewandte
Chemie
Research Articles
12187Angew.Chem. Int.Ed. 2020,59,12186 –12191 T2020 The Authors. Published by Wiley-VCH Verlag GmbH &Co. KGaA, Weinheim www.angewandte.org
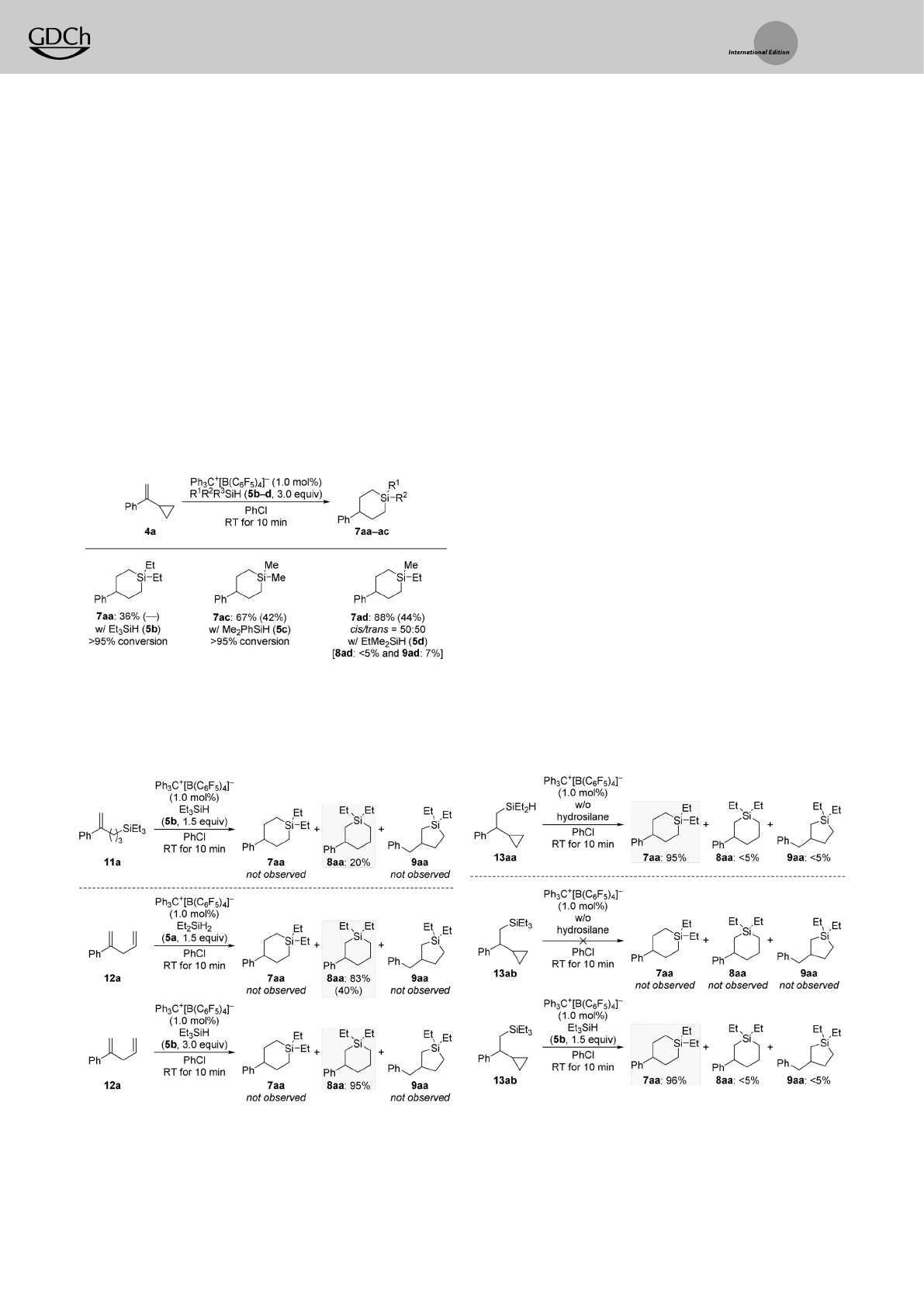
self-regeneration of silylium ions.[7,9,10] Thereactions sum-
marized in Scheme 3demonstrate the feasibility of the
approach. Et3SiH (5b)yielded the same product 7aa as
Et2SiH2(5a)did in the reaction with 4a.Inturn, Me2PhSiH
(5c)underwent preferential dearylation, and EtMe2SiH (5d)
showed that demethylation is favored over abstraction of an
ethyl group.The observations are in line with those previously
made.[14,15]
To gain insight into the reaction mechanism, aseries of
control experiments was designed (Schemes 4–6). We had
recently shown that silylium ions promote the ring-opening
hydrosilylation of cyclopropanes.[7] If the (5++1) cycloaddition
begins with chemoselective ring opening of the cyclopropyl
group in 4,instead of the generation of the cyclopropyl-
stabilized carbenium-ion intermediates 6(cf.Scheme 1,
bottom), the a-substituted styrene derivatives 11 will be
likely intermediates.Hence,weprepared 11a and subjected it
to the standard procedure (Scheme 4, top). The endo
cyclization did occur but without migration of the phenyl
group,and 8aadid form exclusively with no trace of 7aa.This
finding makes the ring-opening preceding functionalization of
the alkene in the VCP unlikely.However,wealso found that
silylium ions can promote the isomerization of acyclopropyl
group into an allyl group.[7] We therefore prepared the 1,4-
diene 12aas another possible intermediate (Scheme 4,
bottom). When reacted with either 5a or 5b it was again
8aa,with no migration of the phenyl group,that was formed
exclusively.[16] These results hint that transposition of the
phenyl group occurs during the ring opening of cyclopropane.
We then turned towards the intermediates 13 with the
cyclopropane ring still intact, that is,the alkene hydrosilyla-
tion products 13aa and 13ab of 4a with 5a and 5b,
respectively (Scheme 5). Theassumed intermediate 13aa,
with aSi
@Hbond, underwent the ring expansion to 7aa
quantitatively with migration of the phenyl group when
treated with 1.0 mol%ofthe trityl borate Ph3C+[B(C6F5)4]@
in the absence of an external hydrosilane (Scheme 5, top).
Thesame result was obtained in the presence of 5b (not
shown). We concluded from this result that intramolecular
hydrosilylation of the cyclopropyl group is interlinked with
the aryl migration. Consequently,repeating this pair of
reactions with presilylated cyclopropane substrate 13ab with
no Si@Hbond led to the expected outcomes (Scheme 5,
bottom). Thetrityl-cation-promoted ring expansion of 13ab
into 7aain the presence of 5blikely involves the dealkylation
of quaternary silanes recently described by us.[14] Hence,
13aa!7aa with no hydrosilane and 13ab!7aa with addi-
tional hydrosilane pass through the same silylium-ion inter-
mediate.
As shown in Scheme 6, aseries of deuterium-labeling
experiments was performed to further elucidate the mecha-
nism. Thedeuterium incorporation was confirmed and
estimated by 1Hand 2HNMR spectroscopy (see the Support-
ing Information for details). When either 4a or the presily-
lated 13ab were reacted with Et3SiD ([D1]5b), deuterium
atoms were found at C3 and C4 of the rearranged product
Scheme 3. Scope II:Variation of the hydrosilane 5in the (5++1)
cycloadditonwith aVCP. Yields determined by 1HNMR spectroscopy
using CH2Br2as an internal standard. Yields of analytically pure
material obtained after flash chromatography on silica gel are given
within parentheses.
Scheme 4. Control experiments I: Verification of ring opening of the
cyclopropyl group in VCPs prior to engagementofthe alkene unit.
Scheme 5. Control experiments II:Cyclization experiments with presily-
lated cyclopropane intermediates, that is, alkene hydrosilylation prod-
ucts of VCPs.
A
ngewandte
Chemie
Research Articles
12188 www.angewandte.org T2020 The Authors. Published by Wiley-VCH Verlag GmbH &Co. KGaA, Weinheim Angew.Chem.Int. Ed. 2020,59,12186 –12191
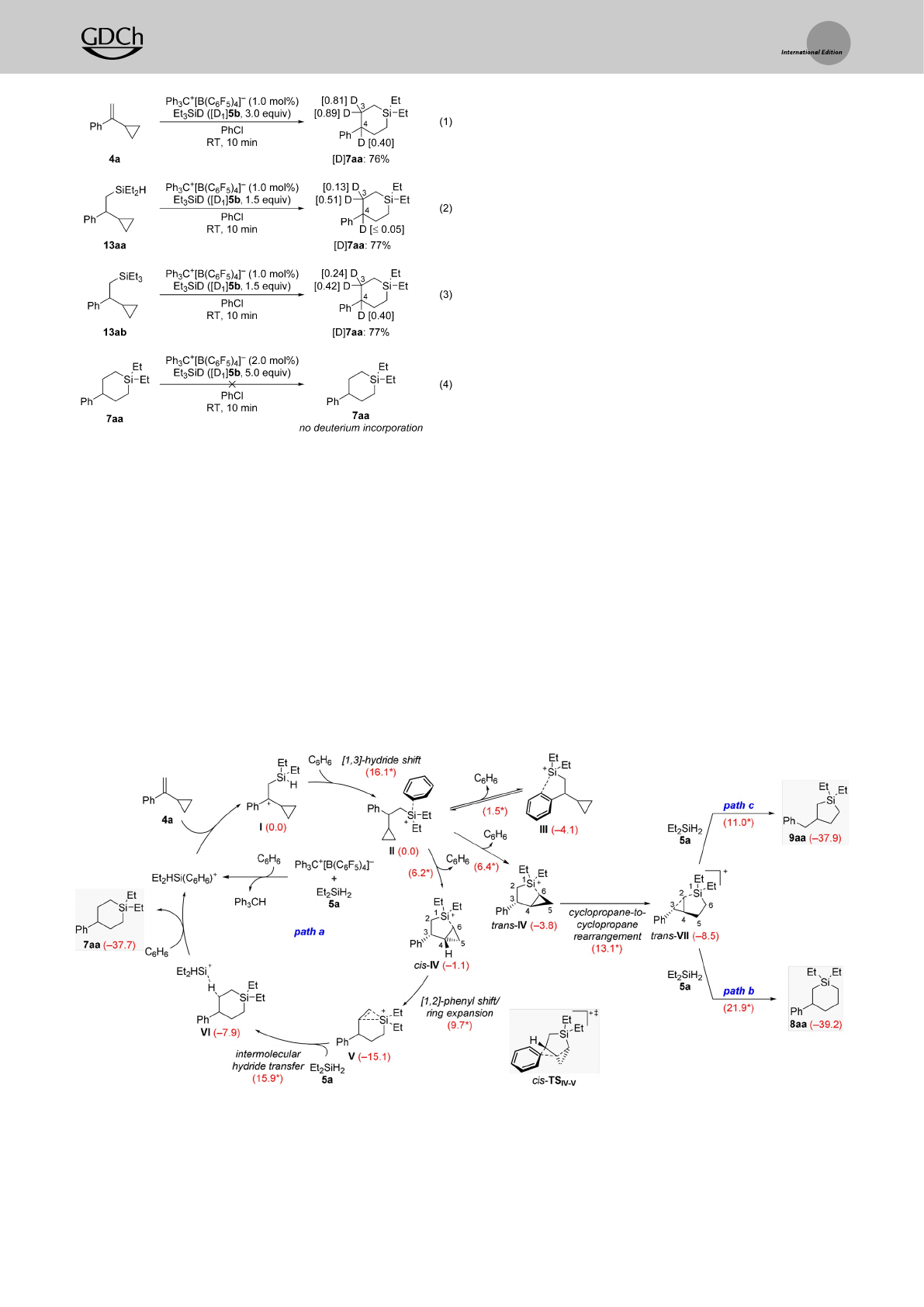
[D]7aa[Eq. (1) and Eq. (3)].Asshown in Scheme 5(top), no
additional hydrosilane is required to convert 13aa into 7aa.
However,when this reaction was performed in the presence
of Et3SiD ([D1]5b), deuterium incorporation was mainly
found at C3 but hardly any at C4 of [D]7aa [Eq. (2)].To
exclude downstream deuteration at C4, we subjected 7aa to
the reaction conditions but did not detect any deuteration in
the benzylic position [Eq. (4)].Consequently,C
@Hbond
formation occurs only during the migration/ring-enlargement
sequence.
To elucidate the mechanistic details of this reaction,
density-functional theory (DFT) calculations at the M062X/
cc-PVTZ//M062X/6–311G(d,p) level[17] were performed on
the model reaction of 4aand 5awith Ph3C+[B(C6F5)4]@as the
initiator (Scheme 7; see the Supporting Information for
computational details and Figures S74–S79 for the free-
energy profile and the optimized structures).[18] Thesolvent
effect was taken into consideration using apolarizable
continuum model (PCM)[19] with benzene as asolvent for
both geometry optimizations and single-point energy calcu-
lations.Benzene was chosen over chlorobenzene because all
byproducts did form in benzene.The initiation step involving
hydride transfer from Et2SiH2to Ph3C+readily occurs over an
activation barrier of 17.4 kcalmol@1.[20] Theresulting hydro-
gen-substituted silylium ion [Et2HSi(benzene)]+can associate
with the C=Cdouble bond in 4a to form the b-silicon-
stabilized carbenium ion Iwith [B(C6F5)4]@as the counter-
anion. This association has been calculated to be exergonic by
16.1 kcalmol@1with respect to the ion pair [Et2HSi-
(benzene)]+[B(C6F5)4]@.Insolution phase,the intermolecu-
larly alkene-stabilized silylium ion IQis predicted to be more
stable than other donor-stabilized silylium ions such as the
corresponding benzene-, chlorobenzene-, hydrosilane-, or
cyclopropyl-stabilized systems (see Tables S1–S3). Therefore,
Iwas selected as the energy reference in the following
discussion. Theion Ithen undergoes an intramolecular
[1,3] hydride shift from the silicon atom to the benzylic
carbon atom to arrive at the benzene-stabilized silylium ion II
over abarrier of 16.1 kcalmol@1(Scheme 7). Subsequent
reorganization of II forms the intramolecularly stabilized
silylium ions III (arene stabilization) or IV (cyclopropane
stabilization in cis-ortrans-configuration), and the corre-
sponding barriers for the formation of these species are 1.5,
6.2, and 6.4 kcalmol@1(relative to II). As aconsequence of the
low free-energy difference between these intermediates,they
Scheme 6. Control experiments III:Deuterium-labeling of the hydro-
silane.
Scheme 7. Initiation and catalytic cycle of the silylium-ion-promoted (5++1) cycloaddition of 4a and 5a (see the SupportingInformation for
calculated structures of relevant intermediatesand transitionstates). Foreach reaction step, the Gibbs free reaction energies and barriers (labeled
with an asterisk) in kcalmol@1were computed with the M06-2X functional.
A
ngewandte
Chemie
Research Articles
12189Angew.Chem. Int.Ed. 2020,59,12186 –12191 T2020 The Authors. Published by Wiley-VCH Verlag GmbH &Co. KGaA, Weinheim www.angewandte.org

are all energetically accessible and likely in equilibrium with
each other.The cyclopropane-stabilized IV can convert into
the b-silicon-stabilized carbenium ion Vthrough aconcerted
[1,2] phenyl shift/ring-expansion transition state (path a). Of
the two configurations of IV,cis-IV gives the lowest
[1,2] phenyl shift/ring-expansion transition state cis-TSIV–V
with asmall activation barrier of 9.7 kcalmol@1(see Fig-
ure S75 for further analysis of structural details of cis-TSIV–V
and trans-TSIV–V).[21] The[1,2] phenyl shift in cis-IV is driven
by the ring expansion of the highly strained bicyclo[3.1.0]hex-
2-silyl cation cis-IV into the six-membered-ring silylium ion
V.[22] Subsequent hydride transfer from 5a to Vaffords the
C(sp3)@H/silylium ion complex VI with abarrier of 15.9 kcal
mol@1.Finally,the association of another molecule of 4awith
Et2HSi+regenerates Iand releases 7aa,that is the major
product obtained experimentally.The [1,3] hydride shift with
an activation barrier of 16.1 kcalmol@1is the rate-limiting step
(I!II), which is consistent with the rapid reaction rate at
room temperature (see Figure S85).
Thedeuterium incorporation in the benzylic position of
7aa (Scheme 6) could be attributed to the formation of the
benzylic cation 7aa+by an intramolecular [1,2] hydride shift
in V(see Figure S84). Such aprocess is endergonic by
4.7 kcalmol@1with an activation barrier of 14.6 kcalmol@1
(relative to V). Further deuteride transfer from [D1]5b to
7aa+forms [D]7aa and regenerates the donor-stabilized
silylium ion. Thecorresponding barrier of this process is
13.9 kcalmol@1.
The[1,2] aryl migration/ring expansion cis-IV!Vof path
aisincompetition with the cyclopropane-to-cyclopropane
rearrangement trans-IV!trans-VII (9.7 versus 13.1 kcal
mol@1;Scheme 7).[21] Theformation of that bicyliccation is
the result of synchronous C2–C4 bond making and C4–C6
bond breaking in trans-IV.Acompeting hydride transfer from
5ato either C4 or C3 of trans-VII furnishes 8aa(path b) and
9aa (path c), respectively.The reaction outcome with 7aa as
the major product and 8aa and 9aa as byproducts is in
accordance with the free-energy difference of those two
barriers (DDG*=3.4 kcalmol@1), making this (5++1) cyclo-
addition akinetically controlled process.
Conclusion
We have disclosed here atransition-metal-free (5++1)
cycloaddition of VCPs and hydrosilanes that is promoted by
the self-regeneration of silylium ions.[7,9,10] Thenew reaction
also involves a[1,2] migration of an aryl group,eventually
furnishing 4- rather than 3-aryl-substituted silacyclohexane
derivatives.Based on various control experiments and
quantum-chemical calculations,reaction mechanisms that
rationalize the formation of the three products have been
proposed. Thebranching point is an intramolecularly cyclo-
propane-stabilized silylium ion that can either undergo
akinetically favored, concerted [1,2] aryl migration/ring
expansion or engage in acyclopropane-to-cyclopropane
rearrangement. That bond reorganization represents
astraightforward and atom-economic access to silacyclohex-
ane derivatives,which are potentially relevant to medicinal
applications.[23]
Acknowledgements
Thework was funded by the Deutsche Forschungsgemein-
schaft (DFG,German Research Foundation) under Germa-
nyQsExcellence Strategy (EXC 2008/1-390540038). G.W.
thanks the China Scholarship Council for apostdoctoral
fellowship (2019–2020). All theoretical calculations were
performed at the High-Performance Computing Center
(HPCC) of Nanjing University.M.O.isindebted to the
Einstein Foundation (Berlin) for an endowed professorship.
Conflict of interest
Theauthors declare no conflict of interest.
Keywords: cycloaddition ·density-functional calculations ·
ring expansion ·silylium ions ·small-ring systems
[1] a) T. Hudlicky,J.W.Reed, Angew.Chem. Int. Ed. 2010,49,
4864–4876; Angew.Chem. 2010,122,4982–4994;b)J.E.
Baldwin, Chem. Rev. 2003,103,1197–1212.
[2] Z. Goldschmidt, B. Crammer, Chem. Soc.Rev. 1988,17,229–
267.
[3] a) G. Fumagalli, S. Stanton, J. F. Bower, Chem. Rev. 2017,117,
9404–9432;b)L.Souillart, N. Cramer, Chem. Rev. 2015,115,
9410–9464;c)Y.Gao,X.-F.Fu, Z.-X. Yu, Top. Curr.Chem.
2014,346,195–231;d)M.Rubin, M. Rubina, V. Gevorgyan,
Chem. Rev. 2007,107,3117–3179;e)S.C.Wang,D.J.Tantillo, J.
Organomet. Chem. 2006,691,4386–4392.
[4] a) G. A. Olah, D. P. Kelly,C.L.Jeuell, R. D. Porter, J. Am.
Chem. Soc. 1970,92,2544–2546;b)G.A.Olah, C. L. Jeuell,
D. P. Kelly,R.D.Porter, J. Am. Chem. Soc. 1972,94,146–156;
c) G. A. Olah, P. W. Westerman, J. Am. Chem. Soc. 1973,95,
7530–7531;d)J.F.Wolf,P.G.Harch, R. W. Taft, W. J. Hehre, J.
Am. Chem. Soc. 1975,97,2902 –2904;e)H.Mayr,G.A.Olah, J.
Am. Chem. Soc. 1977,99,510 –513;f)K.B.Wiberg,D.Shobe,
G. L. Nelson, J. Am. Chem. Soc. 1993,115,10645–10652.
[5] I. Fleming, Chem. Soc.Rev. 1981,10,83–111.
[6] a) J. B. Lambert, Y. Zhao,R.W.Emblidge,L.A.Salvador,X.
Liu, J.-H. So,E.C.Chelius, Acc.Chem. Res. 1999,32,183–190;
for asummary of the chemistry of silyl-substituted carbocations,
see:b)H.-U.Siehl, T. Mgller in The Chemistry of Organic
Silicon Compounds,Part 2 (Eds.: Z. Rappoport, Y. Apeloig),
Wiley,Chichester, 1989,pp. 595–701.
[7] A. Roy,V.Bonetti, G. Wang,Q.Wu, H. F. T. Klare,M.
Oestreich, Org.Lett. 2020,22,1213–1216.
[8] J. Y. Corey, J. Am. Chem. Soc. 1975,97,3237–3238.
[9] a) J. B. Lambert, Y. Zhao,H.Wu, J. Org.Chem. 1999,64,2729 –
2736;b)V.J.Scott, R. C¸ elenligil-C¸etin, O. V. Ozerov, J. Am.
Chem. Soc. 2005,127,2852–2853;c)R.Panisch, M. Bolte,T.
Mgller, J. Am. Chem. Soc. 2006,128,9676–9682;d)K.Mgther,
M. Oestreich, Chem. Commun. 2011,47,334–336;e)K.
Mgther,J.Mohr, M. Oestreich, Organometallics 2013,32,
6643–6646;f)O.Allemann,S.Duttwyler, P. Romanato,K.K.
Baldridge, J. S. Siegel, Science 2011,332,574 –577;g)B.Shao,
A. L. Bagdasarian, S. Popov,H.M.Nelson, Science 2017,355,
1403–1407.
A
ngewandte
Chemie
Research Articles
12190 www.angewandte.org T2020 The Authors. Published by Wiley-VCH Verlag GmbH &Co. KGaA, Weinheim Angew.Chem.Int. Ed. 2020,59,12186 –12191
Loading more pages...