
Bioorthogonal Chemistry Hot Paper
DFT-Guided Discovery of Ethynyl-Triazolyl-Phosphinates as
Modular Electrophiles for Chemoselective Cysteine Bioconjugation
and Profiling
Christian E. Stieger, Yerin Park, Mark A. R. de Geus, Dongju Kim, Christiane Huhn,
J. Sophia Slenczka, Philipp Ochtrop, Judith M. Müchler, Roderich D. Süssmuth,
Johannes Broichhagen, Mu-Hyun Baik,* and Christian P. R. Hackenberger*
In memory of Professor Ulf Diederichsen
Abstract: We report the density functional theory
(DFT) guided discovery of ethynyl-triazolyl-phosphi-
nates (ETPs) as a new class of electrophilic warheads for
cysteine selective bioconjugation. By using CuI-catalysed
azide alkyne cycloaddition (CuAAC) in aqueous buffer,
we were able to access a variety of functional electro-
philic building blocks, including proteins, from diethyn-
yl-phosphinate. ETP-reagents were used to obtain
fluorescent peptide-conjugates for receptor labelling on
live cells and a stable and a biologically active antibody-
drug-conjugate. Moreover, we were able to incorporate
ETP-electrophiles into an azide-containing ubiquitin
under native conditions and demonstrate their potential
in protein–protein conjugation. Finally, we showcase the
excellent cysteine-selectivity of this new class of electro-
phile in mass spectrometry based, proteome-wide cys-
teine profiling, underscoring the applicability in homo-
geneous bioconjugation strategies to connect two
complex biomolecules.
Introduction
The chemical functionalization of proteins allows probing
and altering their biological function and generating new
biotherapeutics, including antibody-drug-conjugates
(ADCs).[1,2] In these applications a homogeneous and well-
defined conjugate is desired.
One approach to achieve this relies on incorporating
unnatural amino acids with distinct bioorthogonal
reactivities.[3–5] Alternatively, one can exploit the inherent
reactivity of canonical amino acid side chains. Chemo-
selective modifications of most amino acids in proteins are
typically carried out by electrophilic reagents targeting
nucleophilic amino acids, especially lysine and cysteine.[2, 6–8]
The sulfhydryl group of cysteine is a particularly attractive
target for bioconjugation due to its low natural abundance
and high nucleophilicity under physiological conditions.[9]
Maleimides are most frequently employed as electrophiles
in cysteine bioconjugation even though the formed thiosuc-
cinimide linkage is susceptible to retro-Michael reaction,
which can be problematic for the conjugation of cytotoxic
payloads.[10,11] To overcome this problem, several alternative
methodologies for cysteine labelling have been developed
including vinylsulfonamides,[12] carbonyl acrylic
derivatives,[13] hypervalent iodine reagents,[14,15] and organo-
metallic reagents.[16,17] Nonetheless, moderate selectivity or
low reactivity under physiological conditions continue to be
problematic.[18]
In addition to selectivity and stability concerns, the
synthetic accessibility and modularity of the electrophile are
important. Whereas peptides can be functionalized with an
[*] C. E. Stieger, Dr. M. A. R. de Geus, C. Huhn, Dr. P. Ochtrop,
J. M. Müchler, Dr. J. Broichhagen, Prof. Dr. C. P. R. Hackenberger
Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP)
Robert-Rössle-Strasse 10, 13125 Berlin (Germany)
E-mail: [email protected]
C. E. Stieger, C. Huhn, Dr. P. Ochtrop, J. M. Müchler,
Prof. Dr. C. P. R. Hackenberger
Department of Chemistry, Humboldt Universität zu Berlin
Brook-Taylor-Straße 2, 12489 Berlin (Germany)
Y. Park, D. Kim, Prof. Dr. M.-H. Baik
Department of Chemistry, Korea Advanced Institute of Science and
Technology (KAIST)
Daejeon 34141 (Republic of Korea)
Y. Park, D. Kim, Prof. Dr. M.-H. Baik
Center for Catalytic Hydrocarbon Functionalizations, Institute for
Basic Science (IBS)
Daejeon 34141 (Republic of Korea)
E-mail: [email protected]
J. S. Slenczka, Prof. Dr. R. D. Süssmuth
Institut für Chemie, Technische Universität Berlin
Strasse des 17. Juni 124, 10623 Berlin (Germany)
© 2022 The Authors. Angewandte Chemie International Edition
published by Wiley-VCH GmbH. This is an open access article under
the terms of the Creative Commons Attribution Non-Commercial
NoDerivs License, which permits use and distribution in any med-
ium, provided the original work is properly cited, the use is non-
commercial and no modifications or adaptations are made.
Angewandte
Chemie
Research Articles www.angewandte.org
How to cite: Angew. Chem. Int. Ed. 2022, 61, e202205348
International Edition: doi.org/10.1002/anie.202205348
German Edition: doi.org/10.1002/ange.202205348
Angew. Chem. Int. Ed. 2022,61, e202205348 (1 of 10) © 2022 The Authors. Angewandte Chemie International Edition published by Wiley-VCH GmbH
electrophile with ease in organic solvents, aqueous environ-
ments pose formidable challenges. Genetically encoding
latent bioreactive electrophiles were partially successful,[19]
but encoding more reactive electrophiles often results in
unspecific side reactions with cytosolic thiols.[20] Therefore,
the most common strategy to position a highly reactive
electrophile on proteins is the use of bis-electrophilic linkers
that can be attached to cysteines or other nucleophilic
residues; nevertheless, undesired intramolecular reactions[21]
and dimerization are often observed.[22,23] Alternatively, a
nucleophilic amino acid can be chemically converted into an
electrophile as exemplified by the umpolung of cysteine,
tyrosine, and histidine.[24–27]
Another attractive application of electrophiles is in the
area of chemoproteomics[28] to label a specific proteome for
convenient separation and profiling. For optimal results, the
labelling reagents should offer a high target coverage, a
uniform modification, excellent amino acid selectivity, and
stability of the formed conjugate.[18] To date, this approach is
widely applied in elucidating the proteome-wide redox-
sensitivity[29] and ligandability of cysteines by electrophilic
fragments,[30,31] natural products,[32] or drugs.[33]
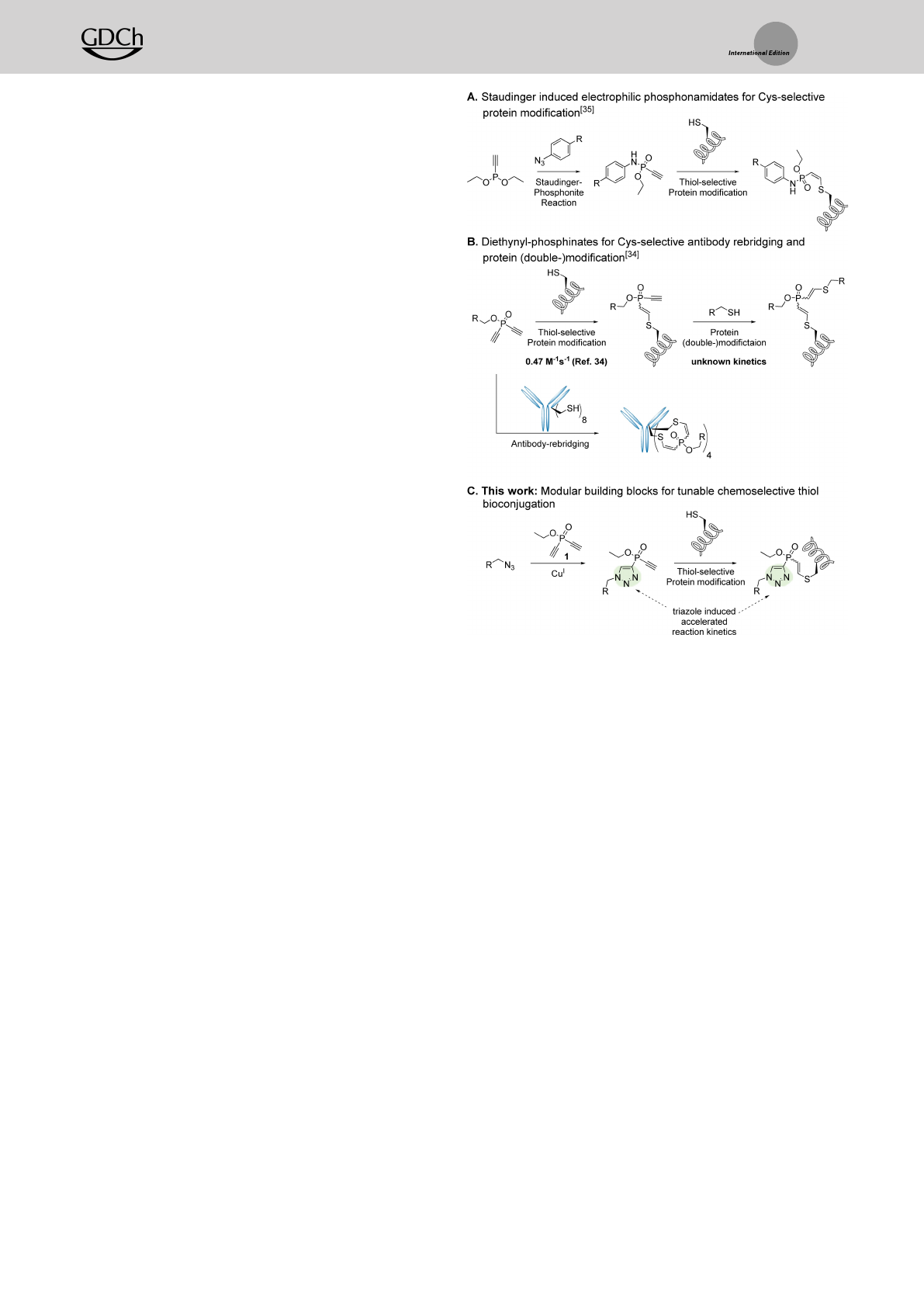
As part of our recent effort to establish diethynyl
phosphinates as disulfide rebridging reagents for stable
antibody modifications, we employed these bisfunctional
cysteine-selective electrophiles for the step-wise functionali-
zation of a protein substrate (Scheme 1B).[34] In this proto-
col, an electrophilic group was installed on a former cysteine
sidechain by reaction with diethynyl phosphinate and further
reacted with a thiol-containing small-molecule or peptide to
generate the desired conjugate. Despite these advances, the
incorporation of thiols into small molecules can be syntheti-
cally laborious and is accompanied by the inherent problem
of disulfide formation. Therefore, we wanted to exploit
other modular and synthetically straightforward strategies to
incorporate cysteine-selective electrophiles into small mole-
cules, peptides and proteins.
Our current paper describes the discovery of 1,2,3-
triazolyl-substituted ethynyl phosphinates (ETPs) as readily
accessible, fast, and highly selective thiol-electrophiles
(Scheme 1C), guided by density functional theory-based
computer models. These molecules can be easily accessed
from various azide-containing molecules via copper-cata-
lyzed azide-alkyne cycloaddition (CuAAC) in aqueous
buffer. Upon cycloaddition, the electrophiles show a
remarkably high reactivity towards cysteine compared to
diethynyl-phosphinates and outperform our previously re-
ported phosphonamidate electrophiles (Scheme 1A)[35] in
antibody-labelling experiments. We showcase the utility of
these reagents in the generation of various biologically
relevant peptide-, protein-, and antibody-conjugates. Addi-
tionally, we demonstrate that diethynyl-phosphinates can be
used to functionalize azide-containing proteins with an ETP-
electrophile in aqueous systems and use it for protein-
protein conjugation. Finally, we demonstrate the excellent
cysteine selectivity of ETP-electrophiles by proteome-wide
reactivity profiling.
Results and Discussion
We started our investigation by determining the kinetic
parameters of the second thiol addition to diethynyl-
phosphinates (Scheme 1B). Thus, we reacted a small thiol-
containing fluorophore (EDANS-SH) with an excess of
ethyl diethynyl-phosphinate 1and obtained thiovinyl-
ethynyl phosphinate 2in good yield (Z-isomer, 76%). 2was
exposed to an equimolar amount of reduced glutathione in
an aqueous buffer at pH 8.5[36,37] and we observed smooth
conversion to a mixture of the E- and Z-isomers of the
glutathione adduct. The second-order rate constant
(0.29 M1s1, Figure S5) was found to be slightly lower than
the kinetics we reported earlier for the first thiol-addition of
the diethynyl-phosphinate (0.47 M1s1, Scheme 1B).[34] To
increase the reaction speed, we first envisioned to use
electron-withdrawing (EWG) alcohol-substituents in 1, as
previously demonstrated for unsaturated
Scheme 1. General concept of unsaturated PV-electrophiles in Cys-
selective protein labelling. A) Electrophilic phosphonamidates, gener-
ated via the chemoselective Staudinger-phosphinonite reaction, react
selectively with cysteine residues on proteins. B) Diethynyl-phosphi-
nates can react selectively with two distinct thiol-nucleophiles and
enable protein double-modification and interchain disulfide rebridging
in IgG antibodies. C) CuI-click functionalisation of diethynyl-phosphi-
nates generates ETP-reagents that show superior reaction kinetics in
cysteine selective protein modification.
Angewandte
Chemie
Research Articles
Angew. Chem. Int. Ed. 2022,61, e202205348 (2 of 10) © 2022 The Authors. Angewandte Chemie International Edition published by Wiley-VCH GmbH

phosphonamidates,[36] however, all attempts to isolate dieth-
ynyl-phosphinates bearing EWG alcohol-substituents failed
due to rapid hydrolysis of the PO bond. Prior work from
our laboratories showed that density functional theory
(DFT) calculations can predict the relative rates of thiol
conjugations to the respective PV-electrophiles.[38] Also,
analysis of the molecular orbitals suggested that the key
factor in enhancing the reactivity is to stabilize the
negatively charged intermediate generated upon the nucleo-
philic attack of thiolate, where delocalization of the anionic
character is achieved through hyperconjugation. The bond
P–XEt (X: S, O, and NH) located in an antiperiplanar
position to the lone-pair played a decisive role in accepting
the electron density. Based on this concept, we interrogated
if we can use the accepting ability of new substituents
featuring π systems accessible from 1(i.e., π-acidity) to
improve the reactivity. We envisioned that several hetero-
cyclic substituents containing electronegative oxygen and/or
nitrogen atoms would be suitable to achieve both, an
electron-withdrawing inductive effect and π-conjugation
with the lone-pair electrons. Therefore, we focused on the
reagents A–Econtaining isoxazole (A), 1,2,3-triazole (Band
C), 1H-pyrazole (D) and 3H-pyrazole (E) substituents and
calculated their reaction profiles using DFT. We modelled
the reactivity of previously reported 2by truncating the
pendant substituent as a methyl group as compound F
(Figure 1).
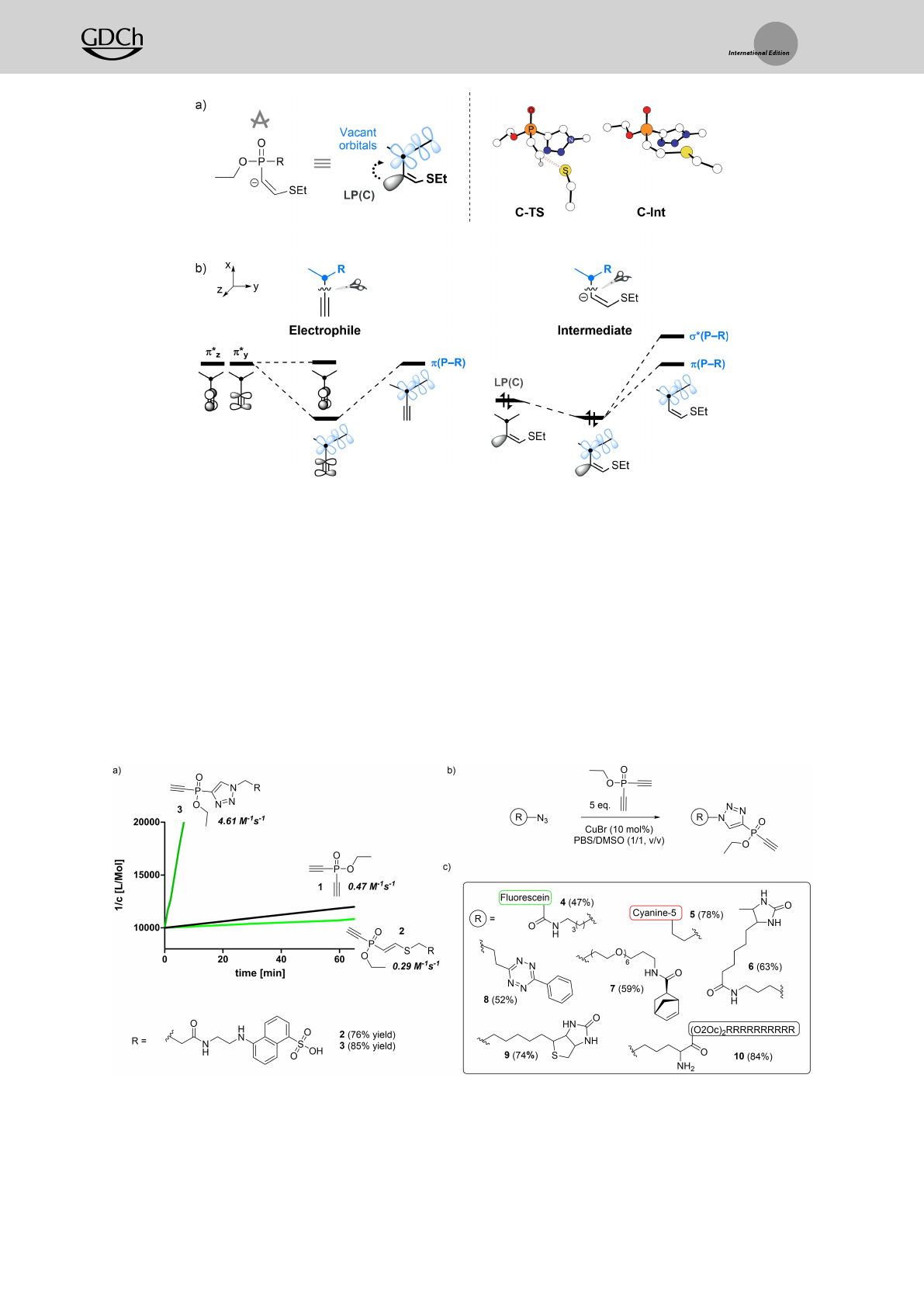
The general scheme of the reaction energy profile for
the Michael-addition to the electrophiles A–Fis shown in
Figure 1, including ethanethiol (EtSH) as a model nucleo-
philic reaction component.
In aqueous buffer (e.g. pH 6.5–8.5), EtSH and the
corresponding thiolate anion EtSare in equilibrium. Since
the thiolate is the better nucleophile, we assumed the
stepwise deprotonation of EtSH and subsequent nucleo-
philic attack of EtSto form the carbanion intermediate.
The calculated reaction barriers (ΔG�) for respective
electrophiles were used to compare the reaction kinetics
among them. Calculated ΔG�values decrease in the order
of 23.3 (F)>20.0 (C)�19.5 (E)�19.0 (D)>18.1 (A)�
18.1 kcalmol1(B), suggesting that the newly designed
electrophiles A–Eshould be much more reactive than F.
Finally, the intermediate is protonated by water irreversibly
to form the final thiol-addition product. The electrophilicity
of the series of PV-reagents can be explained by DFT-
calculated partial charges of the reactive terminal carbon
atom on the ethynyl group, as atomic charges can be used as
a measurement of the electron-withdrawing ability of
various functional groups.[39–43] Natural population analysis[44]
was conducted to compare the inductive effect posed by the
distinctive substituents in electrophiles A,C, and F(Fig-
ure 2). The atomic charges (qC) are correlated to the
reaction barriers, being most positive for A(0.092),
followed by C(0.108), and most negative for F(0.122).
Along with the partial charges, the frontier molecular
orbitals can be used to account for the electrophilicity or
nucleophilicity,[42,45–48] where the energy levels of the orbitals
affect the efficiency of the bond-forming interaction.[49]
Here, the energy of the reactive π* orbital of the PV-
electrophiles is lowest for A(1.65 eV), followed by C
(0.93 eV), and highest for F(0.33 eV), in line with the
reactivity trend (Figure 2).
We further interrogated how the substituents influence
the π-system to enhance the reactivity. Distortion of the
PC=C bond in the geometry of the transition state (TS)
and carbanion intermediate (Int) is characterized by the
preference for the sp2-hybridised lone-pair to be antiperipla-
nar to the PR bond (Figure 3a). The optimized TS and
intermediate structures of C(C-TS and C-Int) are illustrated
in Figure 3a. This structural feature is also found in the
previous system, where PRσ-bonds (R: S, O, and NH)
accept electron density via hyperconjugation. Similarly,
delocalization of electrons into vacant orbitals related to the
PR bonds is feasible as conceptualized in Figure 3b. The
mixing between π-systems of the ethynyl moiety and PR
bond is clearly captured in the LUMO of the electrophiles.
The conjugation renders the π*yorbital more reactive
compared to π*zorbital, except in the case of F, and
ultimately favors the lone-pair electrons to form in that
direction. Following the guideline provided by the theoret-
Figure 1. Reaction energy profile of the thiol addition of the proposed
reagents A–Eand a model compound F. Energies in kcalmol1,
calculated with B3LYP-D3/cc-pVTZ(-f)//B3LYP-D3/6-31G** level of
theory.
Figure 2. Comparison between electrophiles A,C, and F.qC: Natural
atomic charge at the reactive carbon, E(π*): Energy of the reactive π*
orbital of the electrophile. (For more details see Figure S3 and S4).
Angewandte
Chemie
Research Articles
Angew. Chem. Int. Ed. 2022,61, e202205348 (3 of 10) © 2022 The Authors. Angewandte Chemie International Edition published by Wiley-VCH GmbH
ical study, we envisioned that 1,2,3-triazole-substituted
phosphinate electrophiles could be easily accessed via CuI-
catalyzed azide-alkyne cycloaddition.
Therefore, we investigated the synthesis of ETP-electro-
philes starting from EDANS-N3and phosphinate 1. While
1–3 equiv phosphinate in the presence of 10 mol% CuBr
resulted mainly in the di-functionalized phosphinate, 5 equiv
phosphinate allowed us to obtain the desired mono-
functionalized molecule 3in 85% yield (Scheme 2a). To
validate the computational results, we performed kinetic
analysis using glutathione as a model thiol as described
before. Phosphinate 3showed accelerated reaction kinetics
of 4.61 M1s1(Figure S5), thus outperforming phosphinate 2
by roughly 15-times, which is in agreement with the DFT-
calculated reaction barriers (Scheme 2a). These findings
inspired us to synthesize various functionalized ETP-electro-
philes (Scheme 2b). Using the described procedure, we were
able to generate ETP-reagents bearing fluorophores (3,4
Figure 3. a) The general conformation of transition states and intermediates viewed from the top (left). Geometry of C-TS and C-Int as
representatives (right). b) Conceptualized orbital mixing between two fragments for the LUMO of electrophiles and the HOMO of intermediates.
(For the actual molecular orbitals, see Figure S1 and S2).
Scheme 2. a) Schematic representation of the experimental thiol addition kinetics of phosphinates 1–3at pH 8.5. b) Synthetic procedure for the
generation of functionalized ETP-electrophiles. c) Functional electrophiles obtained via the synthetic procedure depicted in b (values represent
isolated yield).
Angewandte
Chemie
Research Articles
Angew. Chem. Int. Ed. 2022,61, e202205348 (4 of 10) © 2022 The Authors. Angewandte Chemie International Edition published by Wiley-VCH GmbH
and 5), affinity-tags (6and 9) or bioorthogonal-click-handles
(7and 8) in moderate to good yield. Moreover, we could
show that this strategy allows for incorporating an ETP-
electrophile into an azide-containing cell-penetrating R10-
peptide (10) (Scheme 2c).
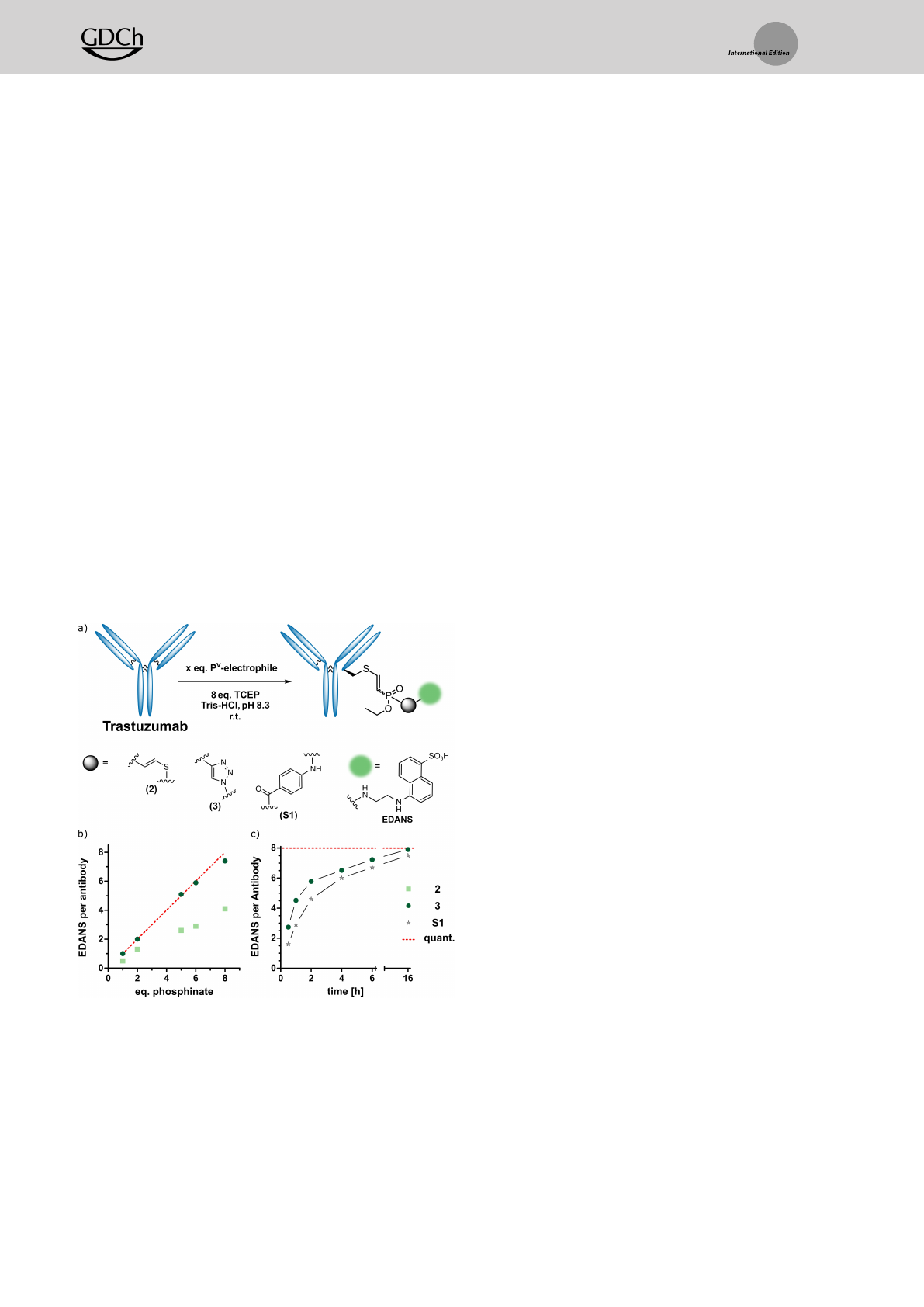
To probe the applicability of ETP-electrophiles for Cys-
selective protein and antibody labelling, we choose the
Her2-targeting therapeutic antibody Trastuzumab as a
model system. At first, we compared thiovinyl-phosphinate
2and triazolyl-phosphinate 3. In brief, Trastuzumab
(5 mgmL1in Tris-buffer pH 8.3) was reduced with 8 equiv
TCEP (37°C, 30 min). Subsequently, 8 equiv of the corre-
sponding phosphinate were added, and the reaction was
allowed to proceed overnight at room temperature (Fig-
ure 4a). While compound 2achieved an average labelling of
4.2 fluorophores per antibody, compound 3reached almost
stoichiometric labelling of all free cysteine thiols in the
antibody corresponding to a fluorophore-to-antibody-ratio
(FAR) of 7.4 (Figure 4b and S6). In a control experiment, in
which the antibody was not reduced prior to the reaction
with the two phosphinates, no modification could be
observed by SDS-PAGE and intact-protein mass spectrom-
etry (MS) (Figure S4b). Also, a larger excess of 3
(100 equiv) did not lead to any antibody-labelling in the
absence of a reducing agent (Figure S7).
The observation of almost stoichiometric labelling when
using 8 equiv EDANS-ETP led us to explore if this is a
general phenomenon. We titrated Trastuzumab with in-
creasing amounts of phosphinates 2and 3(Figure 4a). To
our delight, we observed full antibody labelling with up to
5 equiv and close to stoichiometric labelling with 6 and
8 equiv for phosphinate 3. In contrast, compound 2reached
only approximately 50% labelling for all equivalents after
the same time (Figure 4b).
Finally, we also compared ethynyl-triazolyl-phosphinates
with our previously reported ethynyl-phosphonamidates
(PA) in antibody labelling.[35] We performed a time-course
experiment, in which reduced Trastuzumab was incubated
with 10 equiv of the corresponding PV-electrophile, monitor-
ing the reaction over 16 h using intact-protein MS. We
observed that both the PA- and ETP-reagents reached close
to full conversion (FAR 7.5 and 7.9, respectively) after
overnight reaction; however, after shorter reaction times
phosphinate 3resulted in a higher degree of functionaliza-
tion (Figure 4c).
Apart from fast reaction kinetics and cysteine-selectivity,
also serum-stability of the linkage is a prerequisite for the
successful application in antibody-drug-conjugates to pre-
vent hazardous off-target effects. To test this, we generated
an antibody-fluorescein conjugate using phosphinate 4(see
Supporting Information 3.5) and incubated it in human
serum for 14 days at 37°C. Gratifyingly, no significant
transfer onto other serum proteins was observed (Fig-
ure S8). In addition, we made use of a fluorescence-
quenching assay to validate the serum stability of thiol-thiol
conjugates formed with diethynyl-phosphinates (see Sup-
porting Information 3.6).[34,35] The quenched FRET-pair F1
was synthesized from phosphinate 3and DABCYL-peptide
P2 (38%, Figure S9a). Incubation of F1 in a physiologic
buffer in the presence of excess small thiols and in human
serum did not show any increase in fluorescence signal,
indicating excellent stability (Figure S9b).
Encouraged by the straightforward accessibility, high
reactivity and stability as cysteine-conjugates we used ETP-
electrophiles in the generation of ADCs.
As a payload, we selected Monomethyl-auristatin E
(MMAE), a potent anti-mitotic drug commonly used as a
cytotoxic payload in the generation of ADCs, which we
previously used in thegeneration of phosphonamidate-linked
ADCs.[50] After HPLC-purification, the ETP-functionalized
drug (11) was obtained in 59% yield after HPLC. (Fig-
ure 5a) With the electrophile modified toxin in hand, we
started to examine its applicability in antibody labelling.
Since the experiments using EDANS-ETP 3showed that
already after 4 hours the majority of the reagent has reacted
(Figure 4c), the reactions were terminated and checked after
that time. Intact-protein MS revealed that already 5 equiv of
the ETP-drug was sufficient to reach an average drug-to-
antibody-ratio (DAR) of approximately 4 after the relatively
short reaction time. Hence, we did a scale-up of the reaction
using 1 mg of Trastuzumab to allow further biological
testing. After purification via size-exclusion-chromatography
(SEC), the functionalized antibody was obtained in 80%
yield (0.8 mg). Analysis via hydrophobic-interaction-chro-
matography (HIC) revealed that most of the antibody-
molecules are functionalized with 3–6 drug molecules
resulting in an average DAR of 4.3 (Figure 5b). The cellular
Figure 4. Generation of antibody-fluorophore-conjugates (AFC) using
different PV-electrophiles a) Reaction principle of the simultaneous
reduction alkylation reaction using phosphinates 2and 3and
phosphonamidate S1 b) Equivalent-screen of the two phosphinates
shows close to quantitative labelling with ETP 3c) Time-course of the
antibody labelling reaction using 10 equiv of the corresponding electro-
phile shows superior reaction kinetics for 3compared to phosphona-
midate S1.
Angewandte
Chemie
Research Articles
Angew. Chem. Int. Ed. 2022,61, e202205348 (5 of 10) © 2022 The Authors. Angewandte Chemie International Edition published by Wiley-VCH GmbH
Loading more pages...