Kreuzkupplung Hot Paper
Synthese von unsymmetrischen Azobenzolen durch
palladiumkatalysierte Kreuzkupplung von siliciummaskierten
Diazenylanionen und (Hetero)arylhalogeniden
Lucie Finck und Martin Oestreich*
Abstract: Das photoschaltbare Motiv von Azobenzolen
nimmt eine bedeutende Stellung in den Lebens- und
Materialwissenschaften ein. Das hält einen steten Bedarf
an deren effizienter Synthese aufrecht, was besonders
für unsymmetrische Derivate gilt. Wir stellen hier eine
allgemeine Strategie zu deren Darstellung vor, bei der in
einer neuartigen C(sp2)N(sp2)-Kreuzkupplung funktio-
nalisierte arylsubstituierte, mit einer Silylgruppe mas-
kierte Diazene als Diazenylpronukleophile eingesetzt
werden. Diese Entsprechungen für recht instabile Diaze-
nylanionen kuppeln unter Palladiumkatalyse mit einer
breitgefächerten Palette an (Hetero)arylbromiden ohne
Verlust von Distickstoff. Die konkurrierende Biarylbil-
dung unter Abspaltung von Distickstoff ist vollständig
unterdrückt. Die Reaktion erfordert nur einen geringen
Überschuss von 1.2 Äquivalenten der Diazenylkompo-
nente. Auf diese Weise werden unzählige Azobenzole
mit elektronenreichen/-armen Arylresten unter ausge-
zeichneter Toleranz gegenüber funktionellen Gruppen
planbar zugänglich gemacht.
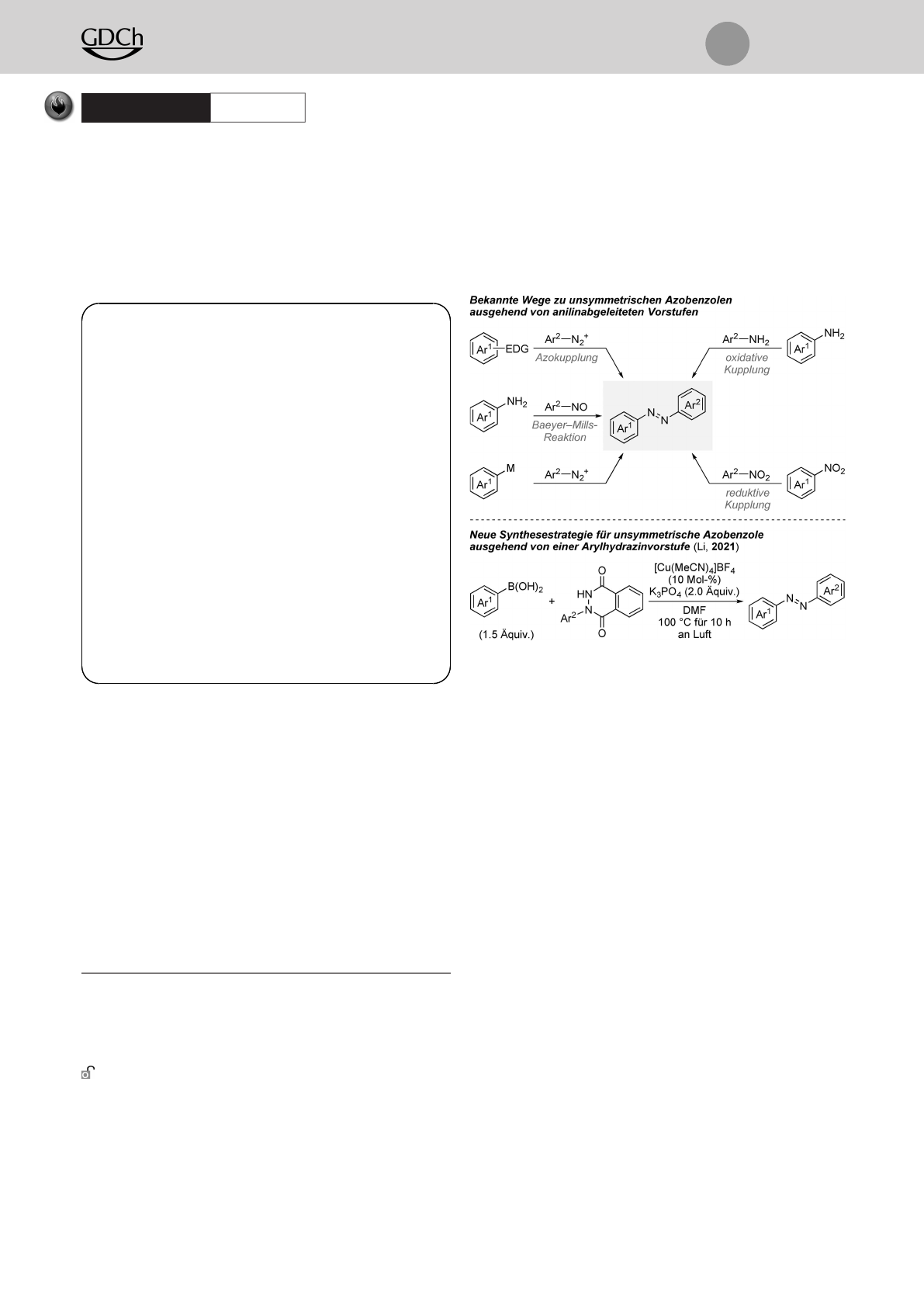
Seit Mitscherlichs fast zweihundert Jahre alter Beschrei-
bung von Azobenzol in der Literatur[1] findet diese Klasse
aromatischer Azoverbindungen wegen ihrer steuerbaren
chemischen und physikalischen Eigenschaften weiterhin
breite Anwendung als organische Farbstoffe, molekulare
Photoschalter und Therapeutika.[2] Brauchbare synthetische
Zugänge zu symmetrisch substituierten Azobenzolderivaten
können gemeinhin als ausgereift angesehen werden, aber
die Darstellung ihrer unsymmetrischen Pendants bleibt eine
Herausforderung (Schema 1, oben).[3] Die Azokupplung ist
eine gängige Strategie, bei der auf eine SEAr-Reaktion eines
Diazoniumsalzes und eines elektronenreichen Aromaten
zurückgegriffen wird.[3,4] Die Kupplung von Nitrosoaroma-
ten und Anilinderivaten unter sauren Reaktionsbedingun-
gen, auch bekannt als die Baeyer–Mills-Reaktion, bietet
einen alternativen Zugang zu unsymmetrischen Produk-
ten.[3,5] Eine hilfreiche Methode, die der eingeschränkten
Anwendungsbreite der oben genannten Reaktionsvorschrif-
ten vorbeugt, ist die Kupplung jener Diazoniumsalze und
metallierter Aromaten.[6,7] Feringa und Mitarbeiter nutzten
dieses Verfahren für die Darstellung von schwer zugängli-
chen tetra-ortho-substituierten, rotverschobenen Azobenzo-
len ausgehend von lithiierten Arylnukleophilen.[7] Azoben-
zole können zudem aus gut verfügbaren und weniger
reaktiven Vorstufen durch dehydrierende oxidative Kupp-
lung von Anilinderivaten[8,9] oder reduktive Heterodimeri-
sierung von Nitroaromaten erhalten werden.[10,11] Obwohl
sich diese Methoden an sich als effizient erwiesen, lässt sich
die Verteilung zwischen homo- und heterodimerisierten
Produkten häufig nicht zufriedenstellend kontrollieren. Um
die bevorzugte Heterodimerisierung zu erreichen, ist norma-
lerweise ein großer Überschuss einer der beiden Kupplungs-
partner vonnöten. Diese Einschränkung wurde jüngst von Li
und Mitarbeitern mit der Entwicklung einer Chan–Evans–
Lam-artigen oxidativen Kreuzkupplung von N-Arylphthal-
säurehydraziden und Arylboronsäuren unter Kupferkatalyse
[*] L. Finck, Prof. Dr. M. Oestreich
Institut für Chemie, Technische Universität Berlin
Straße des 17. Juni 115, 10623 Berlin (Deutschland)
E-mail: [email protected]
Homepage: http://www.organometallics.tu-berlin.de
© 2022 Die Autoren. Angewandte Chemie veröffentlicht von Wiley-
VCH GmbH. Dieser Open Access Beitrag steht unter den Be-
dingungen der Creative Commons Attribution License, die jede
Nutzung des Beitrages in allen Medien gestattet, sofern der ur-
sprüngliche Beitrag ordnungsgemäß zitiert wird.
Schema 1. Syntheserouten zu unsymmetrischen Azobenzolderivaten
mittels CN- oder NN-bindungsknüpfenden Reaktionen. Ar=Aryl-
gruppe, EDG=elektronenschiebene Gruppe, M=Metall.
Angewandte
Chemie
Zuschriften www.angewandte.org
Zitierweise: Angew. Chem. Int. Ed. 2022, 61, e202210907
Internationale Ausgabe: doi.org/10.1002/anie.202210907
Deutsche Ausgabe: doi.org/10.1002/ange.202210907
Angew. Chem. 2022,134, e202210907 (1 of 6) © 2022 Die Autoren. Angewandte Chemie veröffentlicht von Wiley-VCH GmbH

in Angriff genommen (Schema 1, unten).[12] Dieser elegante
Lösungsansatz lieferte eine Unterklasse an Azobenzolen in
moderaten bis guten Ausbeuten.
Vor Kurzem entdeckte unsere Arbeitsgruppe einfach
zugängliche arylsubstituierte Diazene, deren eines Ende
einen Silylrest trägt, wieder.[13] Diese N-Aryl-N’-silyldiazene
sind kinetisch stabil und kamen bereits unter Verlust von
Distickstoff als Arylpronukleophile zum Einsatz.[14,15] Wir
fragten uns, ob dieselben silylierten Diazene ohne Abspal-
tung von Distickstoff auch als Vorläufer von Diazenylanio-
nen fungieren können (Schema 2, oben). Von Diazenen
wird angenommen, dass sie Zwischenstufen bei der Bildung
von Biarylen in palladiumkatalysierten Kreuzkupplungsre-
aktionen von Arylhydrazinderivaten und Arylhalogeniden
unter Freisetzung von Distickstoff sind.[16] Dagegen erhoff-
ten wir uns die direkte Synthese von unsymmetrischen
Azobenzolderivaten ausgehend von den besagten maskier-
ten, arylsubstituierten Diazenen und verschiedenartigen
(Hetero)arylhalogeniden als Kupplungspartner (Schema 2,
unten). Diese unbekannte Kreuzkupplung unterscheidet
sich von dem üblichen Umweg, bei dem auf eine Buchwald–
Hartwig-artige Kreuzkupplung von geschützten Arylhydra-
zinen ein Oxidationsschritt folgt.[17] Wir zielten hingegen auf
eine C(sp2)N(sp2)-Kreuzkupplung von in situ erzeugten
Diazenylanionen und (Hetero)arylelektrophilen unter Palla-
diumkatalyse ab. Dieser seltene Reaktionstyp fand bislang
nur mit Iminen, die als Platzhalter für Ammoniak dienen,
Anwendung.[18,19]
Die oben erwähnte Arbeit von Cho und Mitarbeitern
zur palladiumkatalysierten Arylierung von Hydrazinderiva-
ten war unser Ausgangspunkt bei der Bestimmung des
Katalysatorsystems (Pd2dba3und dppf mit Cs2CO3als Base
und Toluol als Lösungsmittel).[17] Die Wahl dieser Reakti-
onsparameter geschah auf der Grundlage unserer früheren
Erfahrung mit der Aktivierung der silylierten Diazene; die
Freisetzung von Distickstoff ist in polaren Lösungsmitteln,
in denen die silaphile Lewis-Base größtenteils gelöst ist,
wahrscheinlicher.[14] Ein Kontrollexperiment in THF als
Lösungsmittel zeigte allerdings, dass weder der Verlust von
Distickstoff (in Spuren) noch die anvisierte Kupplung ablief;
stattdessen wurde die Defunktionalisierung des Arylbromids
nachgewiesen. Cs2CO3in Toluol erschien uns vielverspre-
chend, und wir legten die Reaktionstemperatur auf 60°C an
Stelle von 110°C fest, um die Abspaltung von Distickstoff
nicht zu begünstigen. Einen ähnlichen Effekt könnte zudem
der große Bisswinkel des dppf-Liganden durch Beschleuni-
gung der reduktiven Eliminierung ausüben.[20] Das para-
tolylsubstituierte Diazenylpronukleophil 1a und das an-
spruchsvollere[21] elektronenreiche Arylbromid 2a wurden
als Modellverbindungen eingesetzt (Tabelle 1; die komplette
Optimierung der Reaktionsbedingungen findet sich in den
Hintergrundinformationen). Jene Reaktionsbedingungen er-
gaben in der Tat das gewünschte Azobenzolderivat 6aa in
guter Ausbeute ohne Bildung des Biaryls 7aa (Nr. 1). Für
einen Wechsel von dem Palladium(0)- zu einem Palladium-
(II)-Präkatalysator führten wir die Kreuzkupplung mit prä-
formiertem (dppf)PdCl2durch. Diese Abwandlung bewirkte
die chemoselektive Bildung von 6aa in 99% Ausbeute nach
15 h (Nr. 2). Abgesehen von der kürzeren Reaktionszeit im
Vergleich zu der mit Chos abgeändertem Verfahren (60°C
an Stelle von 110°C) blieb die Bildung von Nebenprodukten
aus, und die chromatographische Abtrennung von dba
entfiel ebenfalls. Andere im Handel erhältliche Palladium-
(II)-Komplexe wie z.B. (dtbpf)PdCl2, (dppe)PdCl2und
(Ph3P)2PdCl2führten entweder zu einer Mischung von 6aa
und 7aa oder zeigten keinen Umsatz (Nr. 3–5). Anschlie-
ßend richteten wir unsere Aufmerksamkeit auf den Einfluss
der Abgangsgruppe am Arylelektrophil 3a–5a. Die Reakti-
vität sowohl des Aryltriflats 3a als auch des Aryliodids 4a
war mit der des Arylbromids 2a vergleichbar, aber die
Ausbeute und die Chemoselektivität waren geringer (Nr. 6
und 7). Ein Absenken der Reaktionstemperatur auf 45°C
verringerte das Ausmaß an Distickstoffabspaltung vollstän-
dig für das Aryltriflat 3a und teilweise für das Aryliodid 4a
(nicht gezeigt). Das weniger reaktive Arylchlorid 5a reagier-
te nicht (Nr. 8). Etliche Basen, von denen eine NSi-
Bindungsspaltung zu erwarten ist,[14] wurden untersucht.
K2CO3und CsF resultierten wegen ihrer geringen Löslich-
keit in niedrigem Umsatz der Ausgangsmaterialien, wohin-
gegen NaOtBu das Produkt 6aa in 63% Ausbeute ergab
(Nr. 9–11). In Abwesenheit einer Base oder eines Präkataly-
sators wurde keine Reaktion beobachtet (Nr. 12 und 13).
Nach der Ausarbeitung geeigneter Reaktionsbedingun-
gen (Tabelle 1, Nr. 2) wandten wir uns der Untersuchung
der Anwendungsbreite zu (Schemata 3–5). Eine Reihe an
funktionalisierten Silyldiazenen 1a–lwurde mit 1-Brom-3-
fluorbenzol (2b) erfolgreich gekuppelt (Schema 3). Damit
war ein Zugang zu den unsymmetrischen, fluorierten aroma-
tischen Azoverbindungen 6ab–lb geschaffen, deren Darstel-
lung sich mit bekannten Verfahren als schwierig erwiesen
hatte (Schema 3; für einen Überblick bekannter und unbe-
kannter Substitutionsmuster siehe Abbildung S1 in den
Hintergrundinformationen). Kupplungsreaktionen von
Aryldiazenen mit einer elektronenschiebenden 4-Methyl-
(wie in 1a) oder 4-Methoxygruppe (wie in 1b) als Substitu-
ent lieferten die entsprechenden Azobenzole 6ab und 6bb
in ausgezeichneten Ausbeuten, und die Stammverbindung
Phenyldiazen 1c reagierte gleichermaßen gut. Halogenato-
me (F in 1d und Cl in 1e) sowie elektronenziehende
Substituenten waren ebenfalls kompatibel, und die Kupp-
Schema 2. Silylierte Aryldiazene als Aryl- bzw. Diazenylanionsynthone
(oben) und geplante Strategie zum Aufbau unsymmetrischer Azoben-
zolderivate durch C(sp2)N(sp2)-Kreuzkupplung (unten). Ar=(Hete-
ro)arylgruppe, Si=Triorganosilyl, X=(Pseudo)halogen.
Angewandte
Chemie
Zuschriften
Angew. Chem. 2022,134, e202210907 (2 of 6) © 2022 Die Autoren. Angewandte Chemie veröffentlicht von Wiley-VCH GmbH
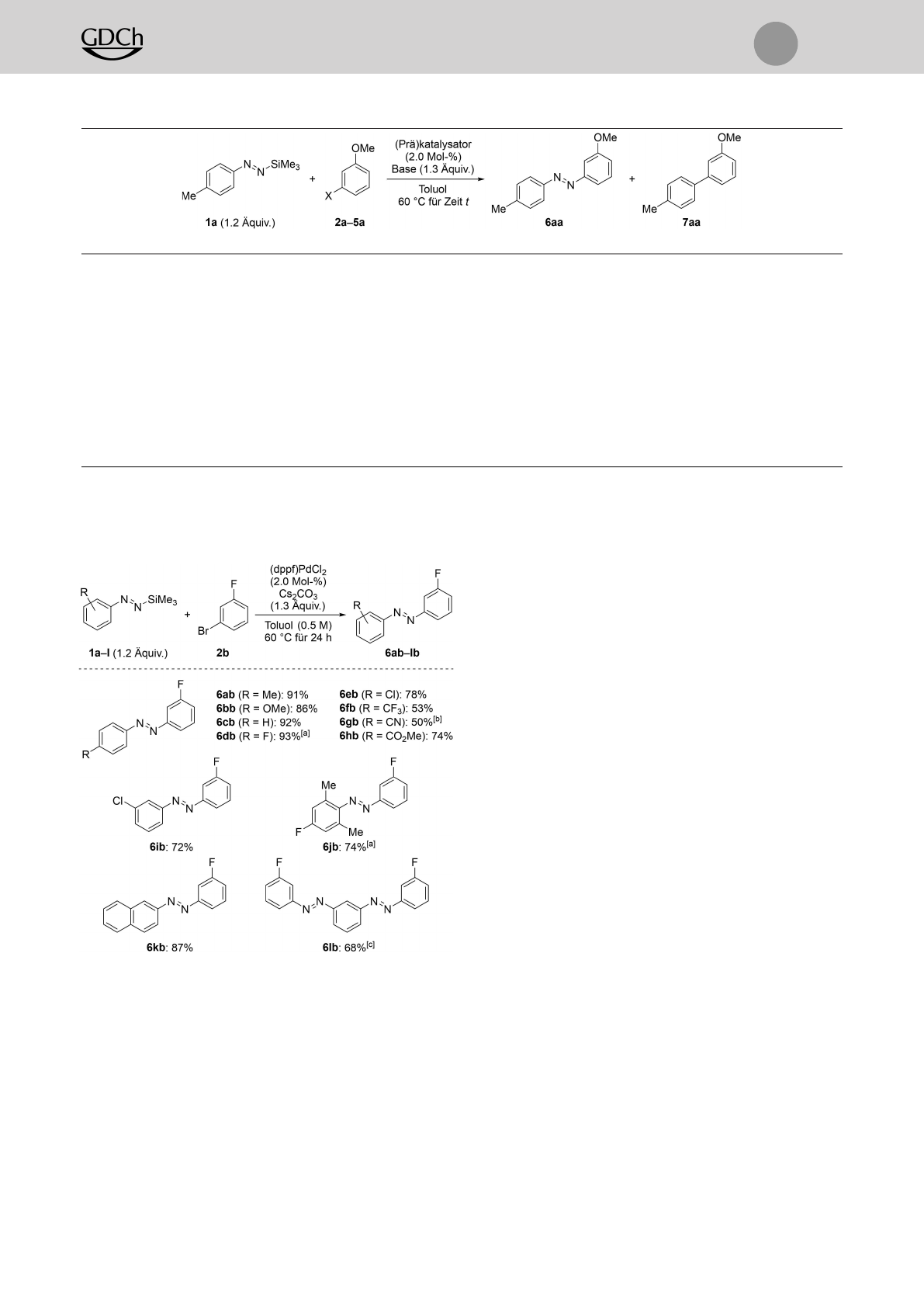
lungsprodukte 6db–hb mit zwei elektronenarmen aromati-
schen Ringen wurden in guten Ausbeuten erhalten. Die
konkurrierende Distickstoffabspaltung und langsamere Re-
aktionsgeschwindigkeiten wurden für Nukleophile, die stark
elektronenziehende Gruppen wie etwa Trifluormethyl (1f)
oder Cyano (1g) tragen, gefunden.[21] Im Einklang damit
zeigte sich das meta-nitrosubstituierte Diazen als unreaktiv
unter den Standardreaktionsbedingungen (nicht gezeigt).
Des Weiteren wirkten sich ortho- und meta-Substituenten
nicht nachteilig aus, und 6ib und 6jb wurden in hohen
Ausbeuten isoliert. Sterisch gehindertes 1j bedurfte einer
herabgesetzten Reaktionstemperatur (45°C), um den Ver-
lust von Distickstoff abzuschwächen. Ein Austausch der
Aryleinheit gegen eine β-Naphthylgruppe war ebenfalls
möglich, und 6kb wurde in 87% Ausbeute erhalten. Ab-
schließend wandten wir unsere Methode auf das 1,3-Bisdia-
zen 1l an, was das Bisazobenzol 6lb in 68% Ausbeute
ergab.
Danach setzten wir eine Auswahl an Arylbromiden
gemäß der optimierten Reaktionsvorschrift um (Schema 4).
Zahlreiche elektronische und sterische Modifizierungen wa-
ren mit unserer Methode vereinbar. Wir testeten zunächst
elektronenreiche Kupplungspartner. (E)-1-(m-Tolyl)-2-(p-
tolyl)diazen (6ac) mit je einer Methylgruppe in einer
bestimmten Stellung an den aromatischen Ringen wurde in
94% Ausbeute isoliert. Außerdem wurde Methoxysubstitu-
tion in der ortho- (2d), meta- (2a) bzw. para-Position (2e)
toleriert. Ebenso führte das dimethylaminosubstituierte
Elektrophil 2f zum Methylgelbderivat 6af, und Brombenzol
(2g) konnte auch eingesetzt werden. Disubstitution in den
ortho- (2h) oder meta-Positionen (2i) lieferte 6ah und 6hi
in hohen Ausbeuten; das gleiche traf auf das catecholabge-
Tabelle 1: Auszug aus der Optimierung der palladiumkatalysierten Kreuzkupplung eines maskierten Diazenylanions und eines elektronenreichen
Aryl(pseudo)halogenids.[a]
Nr. X (Prä)katalysator Base t[h] Ausbeute an 6aa [%][b] Ausbeute an 7 aa [%][b]
1 Br (2a) Pd2dba3/dppf[c] Cs2CO348 90 0
2 Br (2a) (dppf)PdCl2Cs2CO315 99 (92)[d] 0
3[e] Br (2a) (dtbpf)PdCl2Cs2CO348 16 35
4[e] Br (2a) (dppe)PdCl2Cs2CO348 Spuren 0
5[e] Br (2a) (Ph3P)2PdCl2Cs2CO348 Spuren 0
6 OTf (3 a) (dppf)PdCl2Cs2CO315 76 9
7 I (4a) (dppf)PdCl2Cs2CO315 59 28
8[e] Cl (5a) (dppf)PdCl2Cs2CO348 0 0
9[e] Br (2a) (dppf)PdCl2K2CO348 Spuren 0
10[e] Br (2a) (dppf)PdCl2CsF 24 26 Spuren
11[e] Br (2a) (dppf)PdCl2NaOtBu 15 63 0
12 Br (2a) (dppf)PdCl2– 48 0 0
13 Br (2a) – Cs2CO348 0 0
[a] Alle Reaktionen wurden im 0.10-mmol-Maßstab in 0.2 mL Toluol (0.5 M) durchgeführt. [b] Bestimmt mittels kalibrierter GLC-Analyse mit
Tetracosan als internem Standard. [c] 1.0 Mol-% an Pd2dba3und 3.0 Mol-% an dppf. [d] Isolierte Ausbeute im 0.20-mmol-Maßstab nach
Aufreinigung durch Flashchromatographie an Kieselgel in Klammern. [e] Unvollständiger Umsatz des Aryl(pseudo)halogenids. dba=Dibenzyli-
denaceton; dppf=1,1’-Bis(diphenylphosphin)ferrocen; dtbpf=1,1’-Bis(di-tert-butylphosphin)ferrocen; dppe=1,2-Bis(diphenylphosphin)ethan.
Schema 3. Anwendungsbreite I: Palladiumkatalysierte Kreuzkupplung
von funktionalisierten silylierten Aryldiazenen 1a–lund 1-Brom-3-
fluorbenzol (2b). Wenn keine anderen Angaben gemacht werden,
wurden alle Reaktionen im 0.20-mmol-Maßstab durchgeführt. Ausbeu-
ten beziehen sich auf isolierte Produkte nach Aufreinigung durch
Flashchromatographie an Kieselgel. [a] Bei 45°C durchgeführt. [b] Die
Reaktionszeit betrug 48 h. [c] Die Reaktion wurde mit 0.44 mmol an 1-
Brom-3-fluorbenzol (2b) und 0.45 Äquiv. des Bisdiazens 1 l unter
Verwendung von 2.0 Mol-% an (dppf)PdCl2und 1.1 Äquiv. an Cs2CO3
in 0.4 mLToluol durchgeführt (Details in den Hintergrundinformatio-
nen).
Angewandte
Chemie
Zuschriften
Angew. Chem. 2022,134, e202210907 (3 of 6) © 2022 Die Autoren. Angewandte Chemie veröffentlicht von Wiley-VCH GmbH
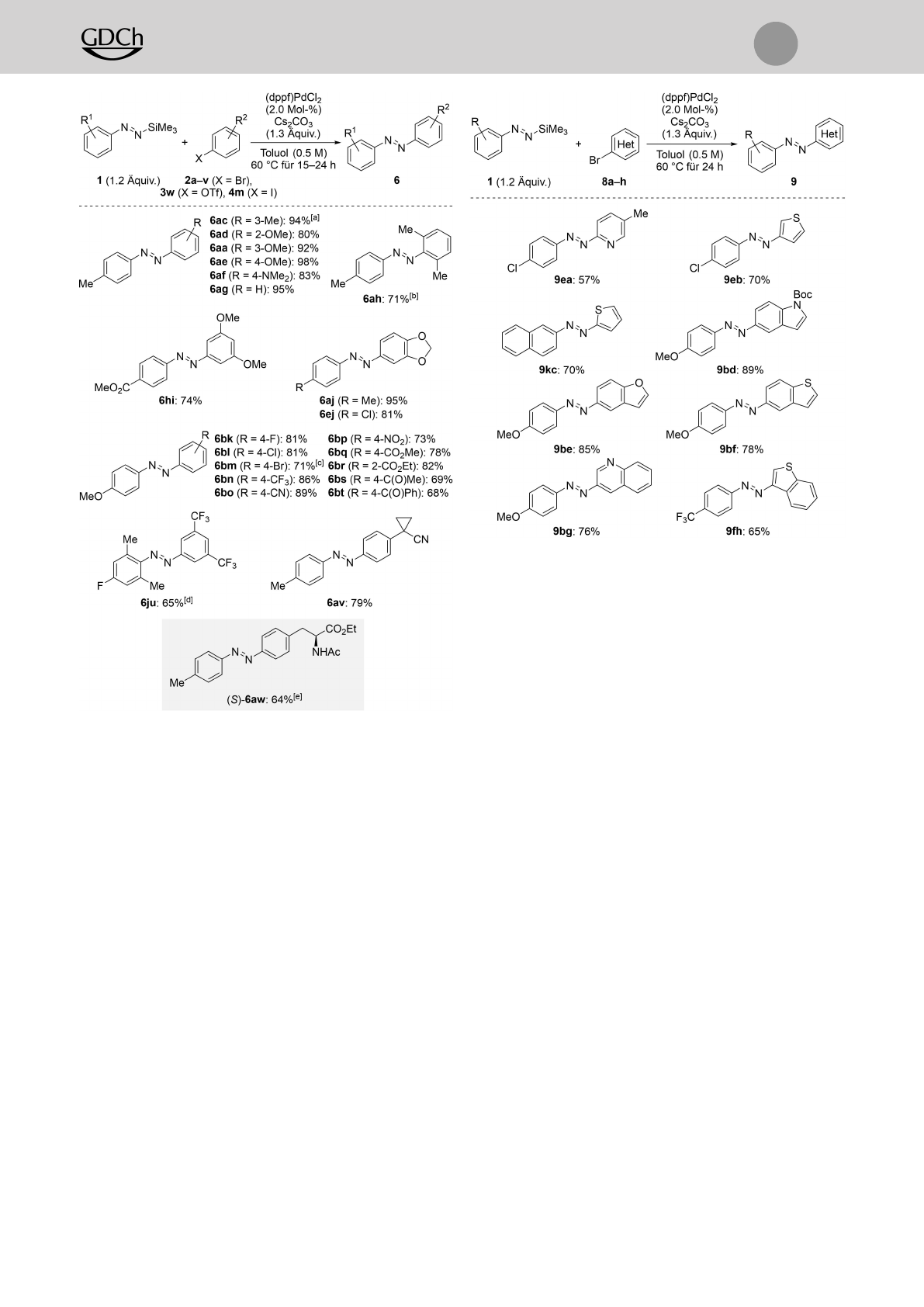
leitete Arylbromid 2j bei der Bildung von 6aj und 6ej zu.
Es ist noch erwähnenswert, dass das sterisch gehinderte 2-
Brom-1,3-dimethylbenzol (2h) nicht unter den Standardre-
aktionsbedingungen reagierte, sich aber bei höherer Reakti-
onstemperatur (80°C) und Katalysatorbeladung (4.0 Mol-
%) zu 6ah umsetzte. Halogene und elektronenziehende
Gruppen wurden gleichermaßen gut geduldet, was die
Darstellung der push-pull-artigen Azobenzolderivate 6bk–
6bt erlaubte. 1-Brom-4-iodbenzol (4m) ging die Kupplung
ausschließlich am iodsubstituierten Kohlenstoffatom unter
Bildung von 6bm ein, ohne dass eine Substitution des
Bromatoms nachweisbar gewesen wäre; mit dem entspre-
chenden Dibrombenzol (2m) wurde hingegen ein unsaube-
res Produktgemisch erhalten. Arylbromide mit empfindli-
chen funktionellen Gruppen wie beispielsweise Nitro (2p),
Alkoxycarbonyl (2q und 2r) und selbst (nicht) enolisierbare
Ketone (2s und 2t) reagierten chemoselektiv in hohen
Ausbeuten. Das sterisch überfrachtete Azobenzol 6ju wurde
bei 45°C in 65% Ausbeute erhalten. Das cyclopropylring-
enthaltende Substrat 2v ergab das gewünschte Kupplungs-
produkt 6av in 79% Ausbeute. Um die Anwendung der
Methode weiter zu veranschaulichen, untersuchten wir am
Ende das von der Aminosäure Tyrosin abgeleitete Aryltri-
flat 3w, welches das Azobenzol (S)-6aw in 64% Ausbeute
erbrachte (grauer Kasten).
Angesichts der Tatsache, dass die Einführung von hete-
roaromatischen Motiven in Azoverbindungen keine einfa-
che Aufgabe ist (siehe Abbildung S1), nahmen wir einige
Heteroarylbromide in unsere Untersuchung auf (Schema 5).
Ohne die allegemeine Reaktionsvorschrift anpassen zu müs-
sen, ging eine Vielzahl heterocyclischer Bromaromaten 8a–
hdie Kupplung in hohen Ausbeuten ein. Azoverbindungen
mit Pyridyl- (wie in 9ea) und Thienyleinheiten (wie in 9eb
und 9kc) wurden in Ausbeuten von 57% bzw. 70% erfolg-
reich isoliert. Wie die Substrate 8b und 8c zeigen, hatte die
Position des Heteroatoms keinen nennenswerten Einfluss
auf die Ausbeute. Benzanellierte Heteroaromaten 8d–hwie
z.B. ein Boc-geschütztes Indol (für 9bd), ein Benzofuran
Schema 4. Anwendungsbreite II: Palladiumkatalysierte Kreuzkupplung
von funktionalisierten silylierten Aryldiazenen 1und verschiedenen
Aryl(pseudo)halogeniden 2a–v,3w und 4 m. Wenn keine anderen
Angaben gemacht werden, wurden alle Reaktionen im 0.20-mmol-
Maßstab durchgeführt. Ausbeuten beziehen sich auf isolierte Produkte
nach Aufreinigung durch Flashchromatographie an Kieselgel. [a] 88%
wurden im 2.0-mmol-Maßstab erhalten. [b] Bei 80°C mit 4.0 Mol-% an
(dppf)PdCl2durchgeführt. [c] 1-Brom-4-iodbenzol (4m) wurde verwen-
det. [d] Bei 45°C durchgeführt. [e] Die Reaktion wurde mit dem
entsprechenden Aryltriflat 3w im 0.10-mmol-Maßstab durchgeführt.
Schema 5. Anwendungsbreite III: Palladiumkatalysierte Kreuzkupplung
von funktionalisierten silylierten Aryldiazenen 1und verschiedenen
Heteroarylbromiden 8a–h. Alle Reaktionen wurden im 0.20-mmol-
Maßstab durchgeführt. Ausbeuten beziehen sich auf isolierte Produkte
nach Aufreinigung durch Flashchromatographie an Kieselgel.
Angewandte
Chemie
Zuschriften
Angew. Chem. 2022,134, e202210907 (4 of 6) © 2022 Die Autoren. Angewandte Chemie veröffentlicht von Wiley-VCH GmbH
(für 9be), ein Benzothiophen (für 9bf und 9fh) sowie ein
Chinolin (für 9bg) besaßen eine hohe Reaktivität und
lieferten die dazugehörigen Heteroazobenzolderivate in
sehr guten Ausbeuten.
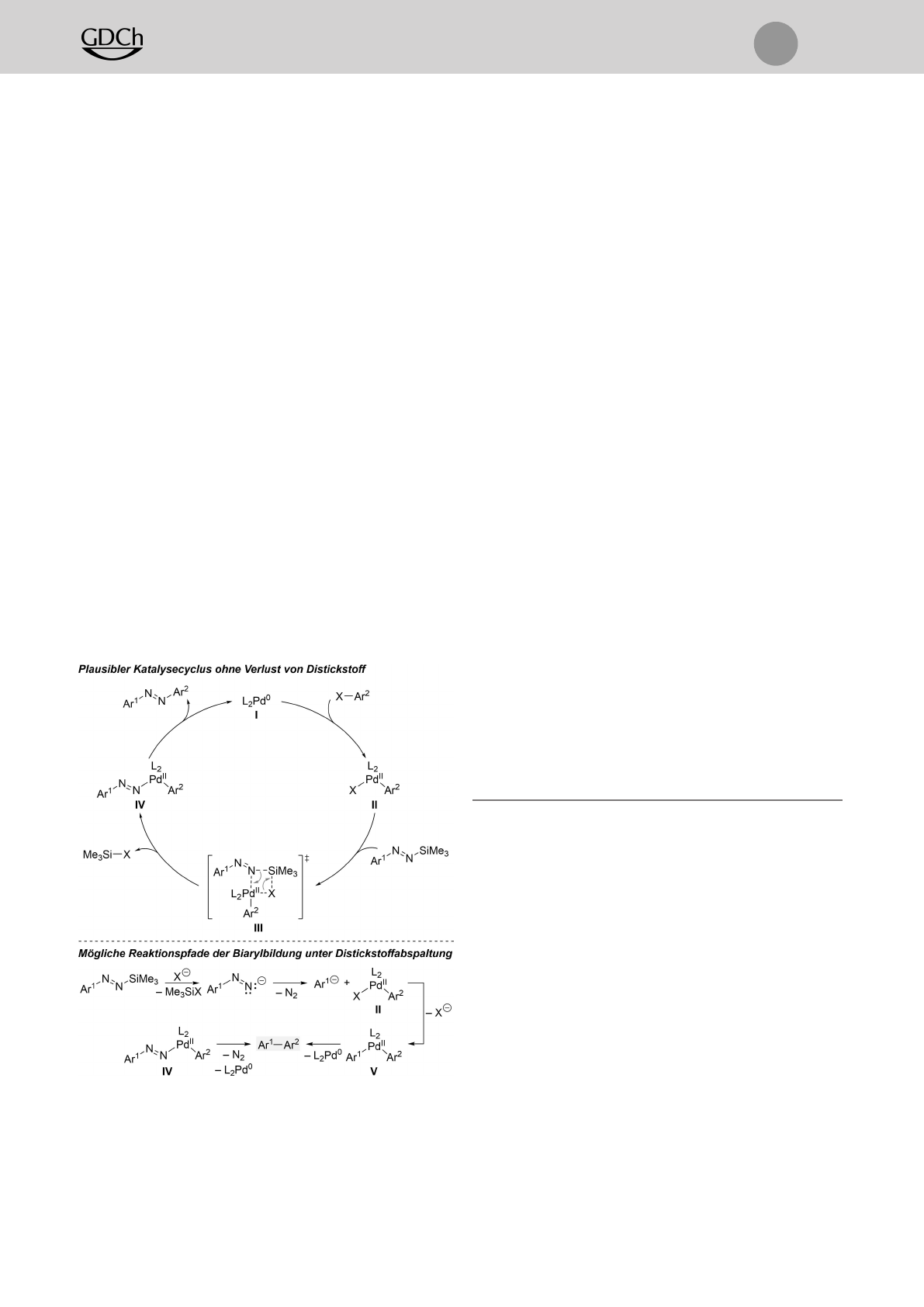
Auf der Grundlage von allgemein akzeptierten Schlüs-
selschritten palladiumkatalysierter Kreuzkupplungsreaktio-
nen[22] wird ein Pd0/PdII-Katalysecyclus vorgeschlagen (Sche-
ma 6, oben). Die neue Arylierung beginnt wahrscheinlich
mit der oxidativen Addition des Arylhalogenids an den
reduzierten Präkatalysator Iunter Bildung des Arylpalladi-
um(II)-Halogenids II. Obwohl Hünig sowie Kosower eigent-
lich schon vor einem halben Jahrhundert gezeigt hatten,
dass das tert-butylsubstituierte Diazenylanion an Carbonyl-
verbindungen addiert werden kann,[23] denken wir nicht,
dass die Transmetallierung mit einem “freien” Diazenylan-
ion einhergeht. Stattdessen ist die Palladium(II)-Zwischen-
stufe II an einer σ-Bindungsmetathese mit dem silylierten
und somit maskierten Diazenylanion beteiligt. Der Über-
gangszustand III setzt dann den Aryl(diazenyl)palladium
(II)-Komplex IV frei, welcher in der Folge eine reduktive
Eliminierung eingeht und so das unsymmetrische Azobenzol
erzeugt. Die Abspaltung von Distickstoff geschieht weder
auf der Stufe von IV noch vor der Transmetallierung, d.h.
III. Diese beiden Reaktionspfade ergäben das ungewollte
Biaryl (grauer Kasten) über die dazu passende Diarylpalla-
dium(II)-Spezies V(Schema 6, unten).[16] Es muss an dieser
Stelle betont werden, dass die Anwesenheit einer Base
ausschlaggebend ist, und sich Cs2CO3als optimal erwiesen
hatte (vgl. Tabelle 1).
Alles in allem entwickelten wir eine effiziente palladium-
katalysierte Kreuzkupplung von Diazenylanionäquivalenten
und (Hetero)aryl(pseudo)halogeniden für den selektiven
Aufbau von unsymmetrischen Azobenzolderivaten. Die
neue Methode erfordert keinen Überschuss eines der beiden
Kupplungspartner, und die Reaktionen werden routinemä-
ßig mit 1.2 Äquivalenten des Diazenpronukleophils durch-
geführt. Das ist auch deshalb möglich, weil unter den
optimierten Reaktionsbedingungen kaum Distickstoff verlo-
ren wird, und daher die Bildung des unerwünschten Biaryl-
produktes nicht konkurriert. Die Vereinbarkeit mit funktio-
nellen Gruppen in beiden Reaktanden ist hervorragend,
wodurch die Synthese von neuen Azobenzolderivaten mit
völlig unterschiedlichen Arylgruppen machbar ist.
Danksagung
Diese Arbeit wurde von der Deutschen Forschungsgemein-
schaft gefördert (Oe 249/23-1). M.O. ist der Einstein Stiftung
Berlin für eine Stiftungsprofessur zu Dank verpflichtet.
Open Access Veröffentlichung ermöglicht und organisiert
durch Projekt DEAL.
Interessenkonflikt
Die Autoren erklären, dass keine Interessenkonflikte vorlie-
gen.
Erklärung zur Datenverfügbarkeit
Die Daten, die die Ergebnisse dieser Studie unterstützen,
sind auf begründete Anfrage beim Autor erhältlich.
Stichwörter: Azoverbindungen ·Chemoselektivität ·
Kreuzkupplung ·Palladium ·Silicium
[1] E. Mitscherlich, Ann. Pharm. 1834,12, 311–314.
[2] Für aktuelle Aufsätze siehe: a) F. A. Jerca, V. V. Jerca, R.
Hoogenboom, Nat. Chem. Rev. 2022,6, 51–69; b) H.-B. Cheng,
S. Zhang, J. Qi, X.-J. Liang, J. Yoon, Adv. Mater. 2021,33,
2007290; c) S. Crespi, N. A. Simeth, B. König, Nat. Chem. Rev.
2019,3, 133–146; d) W. Szymański, J. M. Beierle, H. A. V.
Kistemaker, W. A. Velema, B. L. Feringa, Chem. Rev. 2013,
113, 6114–6178; e) A. A. Beharry, G. A. Woolley, Chem. Soc.
Rev. 2011,40, 4422–4437; f) M.-M. Russew, S. Hecht, Adv.
Mater. 2010,22, 3348–3360; g) Industrial Dyes: Chemistry,
Properties, Applications (Hrsg.: K. Hunger), Wiley-VCH,
Weinheim, 2003.
[3] Für Aufsätze siehe: a) S. S. Kurup, S. Groysman, Dalton Trans.
2022,51, 4577–4589; b) E. Merino, Chem. Soc. Rev. 2011,40,
3835–3853; c) K. Rück-Braun, S. Dietrich, S. Kempa, B. Prie-
wisch, in Science of Synthesis: Houben–Weyl Methods of
Molecular Transformations, Bd. 31b (Hrsg.: C. A. Ramsden, D.
Bellus), Thieme, Stuttgart, 2007, S. 1425–1537.
[4] Für die grundlegende Arbeit zur Azokupplung siehe: P. Griess,
Justus Liebigs Ann. Chem. 1858,106, 123–125.
Schema 6. Vorgeschlagener Katalysecyclus (oben) und konkurrierende
Pfade unter Verlust von Distickstoff (unten). Ar=(Hetero)arylgruppe,
X=(Pseudo)halogen.
Angewandte
Chemie
Zuschriften
Angew. Chem. 2022,134, e202210907 (5 of 6) © 2022 Die Autoren. Angewandte Chemie veröffentlicht von Wiley-VCH GmbH
Loading more pages...